
摘要
在醫療器材產業中,品質管理系統已成為國際買主選擇製造合作夥伴的重要依據。對於骨科植入物的開發與代工製造而言,是否具備ISO 13485認證,往往是洽談初期就被確認的關鍵條件。本文從製造廠的實務角度出發,解析ISO 13485在醫療器材製造中的核心價值、全球法規連結、PDCA循環實踐,以及製造商在導入過程中常見的挑戰與解決方案。文章適合醫材品牌的採購主管、研發人員、品質管理人員,以及正在評估OEM合作夥伴的決策者參考,協助您在供應鏈選擇、法規遵循與品質保證之間找到最佳平衡點。
Table of Contents
1.前言:從價格導向到品質導向的產業轉型
隨著全球法規逐步強化,醫療器材產業正從價格導向轉向品質與製程穩定性的全面提升。這樣的變化源於臨床安全意識提高與各國監管要求升級,不論是歐盟 MDR、FDA QMSR,或亞洲各市場的新規,皆持續強調製造過程必須具備可追溯、可驗證與可重複的一致性能力。
產業研究指出,醫療器材召回事件中約有七成以上源自製造品質控管,而非設計缺陷。這反映出一個重要現實,即使產品設計再具創新或臨床優勢,只要製程管理不穩定,量產後仍可能出現尺寸偏差、表面品質落差、材料批次差異或檢驗疏漏,進而影響病患安全與企業信任度。
因此,全球品牌在選擇 OEM 合作夥伴時,重點早已不在於「能否加工」,而是「能否長期、穩定、可證明地做到品質一致」。ISO 13485 在此扮演關鍵角色。它不只是一張證書,而是製造商是否具備風險管理、文件化流程、設備維護、製程監控、檢驗與追溯能力的重要指標,也是國際品牌評估供應鏈風險的核心依據。
在骨科植入物領域,品質穩定性的重要性更被放大。脊椎固定系統、創傷骨科產品與顱顏重建植入物需長期留置人體並承受反覆應力,任何製程中的微小變異,都可能成為臨床風險。國際品牌因此特別重視製造端是否具備一致的加工參數控制、可追溯的材料來源管理、穩定的表面處理流程與完善的檢驗與紀錄架構。
在這樣的產業背景下,建立一套符合 ISO 13485 並確實落實於日常生產的品質管理系統,不再是加分項,而是進入全球醫療器材市場的基本門檻。它同時代表製造商的可靠度、可預測性與風險控管能力,也是邁向國際合作所不可或缺的專業基礎。
2.ISO 13485的核心價值:不只是認證,更是管理哲學
2.1 什麼是 ISO 13485
ISO 13485 是一項專為醫療器材產業設計的國際品質管理系統標準,目的在協助製造商、供應商與相關服務提供者,建立一套能持續符合法規要求並確保產品安全與一致性的管理架構。此標準涵蓋設計、開發、生產、安裝到售後服務,已成為全球醫療器材產業最重要的遵循基礎之一。
相較於通用的 ISO 9001,ISO 13485 更聚焦於產品安全、法規遵循、風險管理與追溯要求。它強調製造商在滿足客戶需求的同時,也必須確保產品在任何時點都符合安全與法規標準。這種「法規優先」的觀念,是醫療器材產業與其他製造業截然不同的關鍵特性。
2.2 從設計到製造的全流程控管
在全球監管越來越嚴格的環境下,ISO 13485 不只是品質認證,更是醫材製造商用來建立可持續、可驗證品質系統的實務指南。其核心價值主要體現在製程穩定性、追溯能力與風險管理三個層面。
2.2.1 製程穩定性
ISO 13485要求製造商建立標準化流程,確保每批產品都能穩定生產並維持一致品質,降低製程變異風險。這包含原料進料、加工參數設定、品質檢驗到最終包裝,每個環節都需要明確的作業標準與控管機制。
在骨科植入物領域,這項要求尤其重要。像脊椎固定螺釘的螺紋精度、表面粗糙度、材料機械性能等,都必須在規格內保持一致。任何批次間的差異,都可能影響臨床表現與使用安全性。

2.2.2 記錄與追溯能力
ISO 13485要求從原料來源、製程參數、檢驗紀錄到出貨流程,都要具備完整且可查核的追溯資料。這是法規要求,也是風險管理的基礎。當產品在市場上接獲反應或可能出現風險時,完整的追溯資料能協助製造商快速辨識受影響的批次並採取矯正措施。
在實務上,這代表每件產品都必須擁有唯一識別碼,能追溯到原料批號、加工設備、操作人員、量測數據等詳細資訊。這樣的追溯架構,是建立國際合作信任與品質透明度的必要元素。

2.2.3 風險管理能力
ISO 13485將風險識別與控管融入產品全生命週期,透過「預防優於檢驗」的方式,從設計起就先消除可能的風險來源並提升法規符合度。
以 Ti-6Al-4V 鈦合金植入物為例,製造商需要評估刀具磨耗是否影響螺紋精度、加工溫度是否改變材料機械性質、表面處理是否影響生物相容性等。透過系統化風險評估,可提前建立控制措施,降低品質事件的可能性。

2.3 供應鏈選擇與競爭力的關鍵
對醫療器材品牌商而言,這些要求是選擇可信賴供應鏈的核心依據;對 OEM 製造商而言,則是取得國際合作信任與建立競爭力的基本門檻。隨著各國法規持續與 ISO 13485接軌,全球醫療器材產業正朝向更系統化與流程導向的模式發展,品質管理也逐漸成為製造端最重要的實力指標。
3. 全球法規與ISO 13485的連結:一套系統,多國適用
3.1 ISO 13485在全球法規體系中的角色
ISO 13485是國際醫療器材產業普遍採用的品質管理標準,已成為各國法規制度設計的重要參考來源。從歐盟 MDR、美國 FDA QMSR,到台灣 TFDA、日本 PMDA,主要監管機構皆以 ISO 13485的架構為基礎,制定符合當地需求的品質管理要求。這代表全球醫療器材監管正逐漸朝同一品質語言靠攏,使企業在不同市場之間切換時具備更高的一致性與透明度。
這樣的全球趨勢也為製造商與品牌商帶來明顯優勢。選擇具備 ISO 13485 認證的合作夥伴,有助於降低法規不符合的風險,加速技術文件準備並縮短審查時程。對希望進軍海外市場的企業而言,這不僅提升了跨國遵循效率,也讓團隊能把更多資源投入在產品創新與市場拓展,強化整體競爭力。
3.2 主要市場的法規對應
|
國家/地區 |
法規名稱 |
法源依據/版本 |
ISO 13485參照情況 |
關鍵特點 |
|
台灣 |
醫療器材品質管理系統查核準則 |
依據《醫療器材管理法》第22條,2021年5月1日生效 |
第2條明文規定參照ISO 13485:2016訂定,作為製造業者QMS查核依據 |
製造許可有效期3年,至2024年4月30日應全面符合 |
|
歐盟 |
醫療器材法規 |
EU MDR 2017/745,2021年5月26日正式實施 |
第10條要求建立QMS,ISO 13485為主要參考標準。EN ISO 13485:2016+A11:2021將MDR要求與ISO結合 |
|
|
美國 |
品質管理系統法規 |
2024年1月31日公告,2026年2月2日全面實施 |
透過「納入引用」方式將ISO 13485:2016納入21 CFR Part 820 |
增加標示與包裝控制(820.45)、UDI等FDA特定要求 |
|
日本 |
依PMD Act實施,2021年3月26日修訂版對應ISO 13485:2016 |
第2章(第5-64條)與ISO 13485:2016對應,第3章規定追加要求 |
需醫療器材檔案(Medical Device File)、有效期5年 |
表1:全球主要市場ISO 13485法規對應表
|
比較項目 |
台灣TFDA |
歐盟MDR |
美國FDA QMSR |
日本PMDA |
|
實施時間 |
2021年5月1日 |
2021年5月26日 |
2026年2月2日 |
2021年3月26日 |
|
過渡期 |
至2024年4月30日全面符合 |
MDD證書有效至2024年5月26日 |
2年(2024-2026) |
3年過渡期 |
|
ISO對應程度 |
完全參照ISO 13485:2016 |
需符合EN ISO 13485:2016+A11:2021 |
納入引用ISO 13485:2016 |
第2章對應ISO 13485:2016 |
|
認證要求 |
必須通過QMS查核取得製造許可 |
需公告機構認證 |
不需ISO證書但需符合QMSR |
需PMDA或RCB的QMS適合性調查 |
|
有效期限 |
3年 |
通常3年 |
持續符合性 |
5年 |
|
特殊要求 |
免取得製造許可品項公告 |
臨床評估、EUDAMED |
標示與包裝控制 |
醫療器材檔案、MAH要求 |
表2:各國法規的關鍵差異比較
3.3 各市場詳細說明
3.3.1 台灣TFDA
台灣的「醫療器材品質管理系統查核準則」(QMS)於2021年4月14日公告,並於2021年5月1日跟隨《醫療器材管理法》一同生效。第2條明文規定本準則內容參照ISO 13485:2016訂定,作為製造業者QMS查核依據。
既有醫療器材製造業者應於2024年4月30日前全面符合QMS準則並取得醫療器材製造許可。製造許可有效期為3年,到期前6至12個月需申請後續查廠。
3.3.2 歐盟MDR
歐盟醫療器材法規(EU MDR 2017/745)自2021年5月26日正式實施,第10條第9款要求製造商建立品質管理系統,其要求與EN ISO 13485:2016高度相似。2021年發布的EN ISO 13485:2016+A11:2021新增附錄ZA和ZB,將MDR和IVDR的要求與ISO 13485標準的具體條文結合。
歐盟的要求特別強調臨床評估、上市後監督(Post-Market Surveillance)與警戒系統(Vigilance System),製造商需要建立完整的產品生命週期管理機制。雖然ISO 13485不是MDR的直接要求,但它是歐盟統一標準清單中唯一列出的QMS標準,符合ISO 13485即視為符合CE標誌的基本品質保證要求。
3.3.3 美國FDA QMSR
美國FDA於2024年1月31日發布最終法規,將品質系統法規(Quality System Regulation, QSR)修訂為品質管理系統法規(Quality Management System Regulation, QMSR),預計2026年2月2日全面實施。QMSR透過「納入引用」(Incorporation by Reference)方式,將ISO 13485:2016納入21 CFR Part 820,完全對應國際標準。
最終法規要求遵守ISO 13485,並增加符合食品、藥物與化妝品法(FD&C Act)所需的額外要求,包括設計控制的適用範圍、特定記錄保存要求、標示與包裝控制(新增820.45條款)等。這項法規的推出,標誌著美國正式與國際標準接軌,對全球醫材製造商而言是重大利多。
3.3.4 日本PMDA
日本的QMS省令(厚生勞動省令第169號, MHLW MO169)於2021年3月26日修訂,以對應ISO 13485:2016,過渡期為3年。QMS省令第2章(第5-64條)與ISO 13485:2016的第4-8條對應,要求幾乎相同但存在一些細微差異。
第3章規定追加要求事項,特別是醫療器材檔案(Medical Device File)的要求,此要求與歐盟MDR的技術文件不同,而更類似於FDA的Device Master Record。日本市場對文件完整性與追溯性要求特別嚴格,持有ISO 13485證書並不能證明符合日本QMS要求,製造商需要滿足省令第169號的追加要求。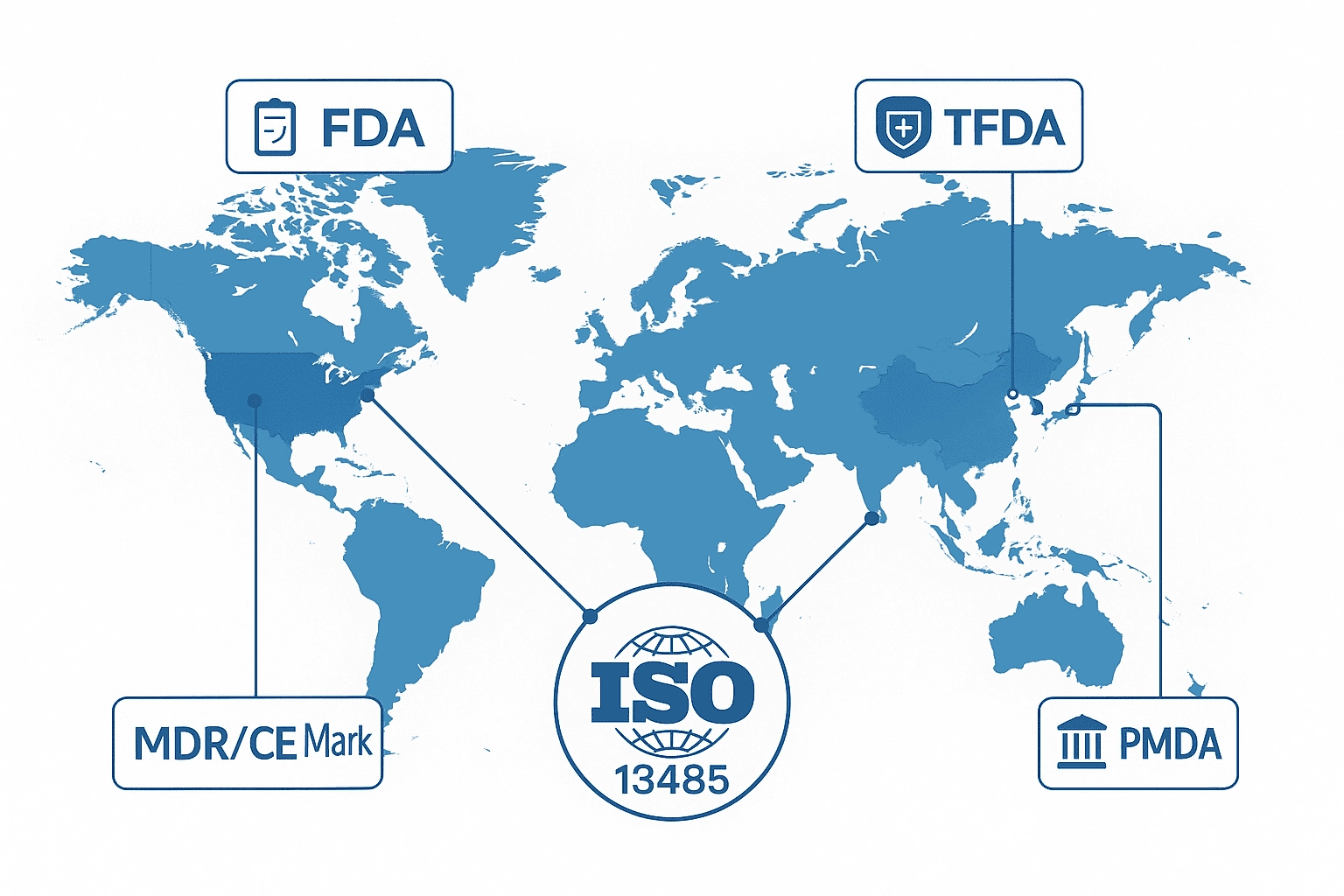
3.4 一套系統應對多國法規的策略
對希望拓展海外市場的醫療器材企業而言,最有效的方式是建立一套「高標準」的 ISO 13485 品質管理系統,並同時滿足主要國家與地區的法規要求。這種做法讓企業在面對各國審查時具備更高的一致性與效率,也能在產品生命週期的每個階段降低遵循風險。
3.4.1 文件管理策略
企業可採用最嚴格的記錄要求作為基準,強化追溯能力並滿足不同市場的需求。建議同時符合 FDA 的 Device History Record、歐盟的技術文件架構以及日本的 Medical Device File 要求,並建立統一的文件管理平台,使其能支援多國法規所需的格式與內容,提升文件維護的一致性與可控性。
3.4.2 風險管理整合
建立系統化的 FMEA 流程,並確保符合 ISO 14971 標準,將風險管理融入產品設計與製造的每個階段。風險評估不應被視為獨立文件,而應成為日常決策與設計調整的依據,使潛在風險能在製程早期被識別與控制,提高產品安全性。
3.4.3 供應鏈品質保證
企業應定期稽核關鍵供應商,並維護合格供應商清單,以確保材料來源與外包製程的穩定性。供應商的品質管理能力會直接影響最終產品的遵循性,因此健全的供應鏈管控是跨國市場策略的核心要素之一。
3.4.4 製程驗證完整性
建立完善的 IQ、OQ、PQ 程序,特別針對滅菌製程、特殊製程與軟體驗證。製程驗證不應僅是取得法規認可的前置作業,而需要透過定期再驗證維持其長期穩定性,確保產品在不同批次間仍能保持同等品質。
3.4.5 上市後監督系統
企業需建立主動式的上市後監督機制,包括不良事件回報、投訴處理、警戒通報與定期安全性更新報告。這些資訊不僅能滿足法規要求,也能作為產品改善、製程更新與風險控制的重要依據,形成品質優化的正向循環。
這種「一次到位」的策略雖然在初期投入較高,但能顯著降低後續進入不同市場時的調整成本,加速產品上市時程。更重要的是,它能強化製造商的核心競爭力,使企業在全球醫療器材市場中建立穩定而長期的優勢。
4. PDCA循環的實際應用
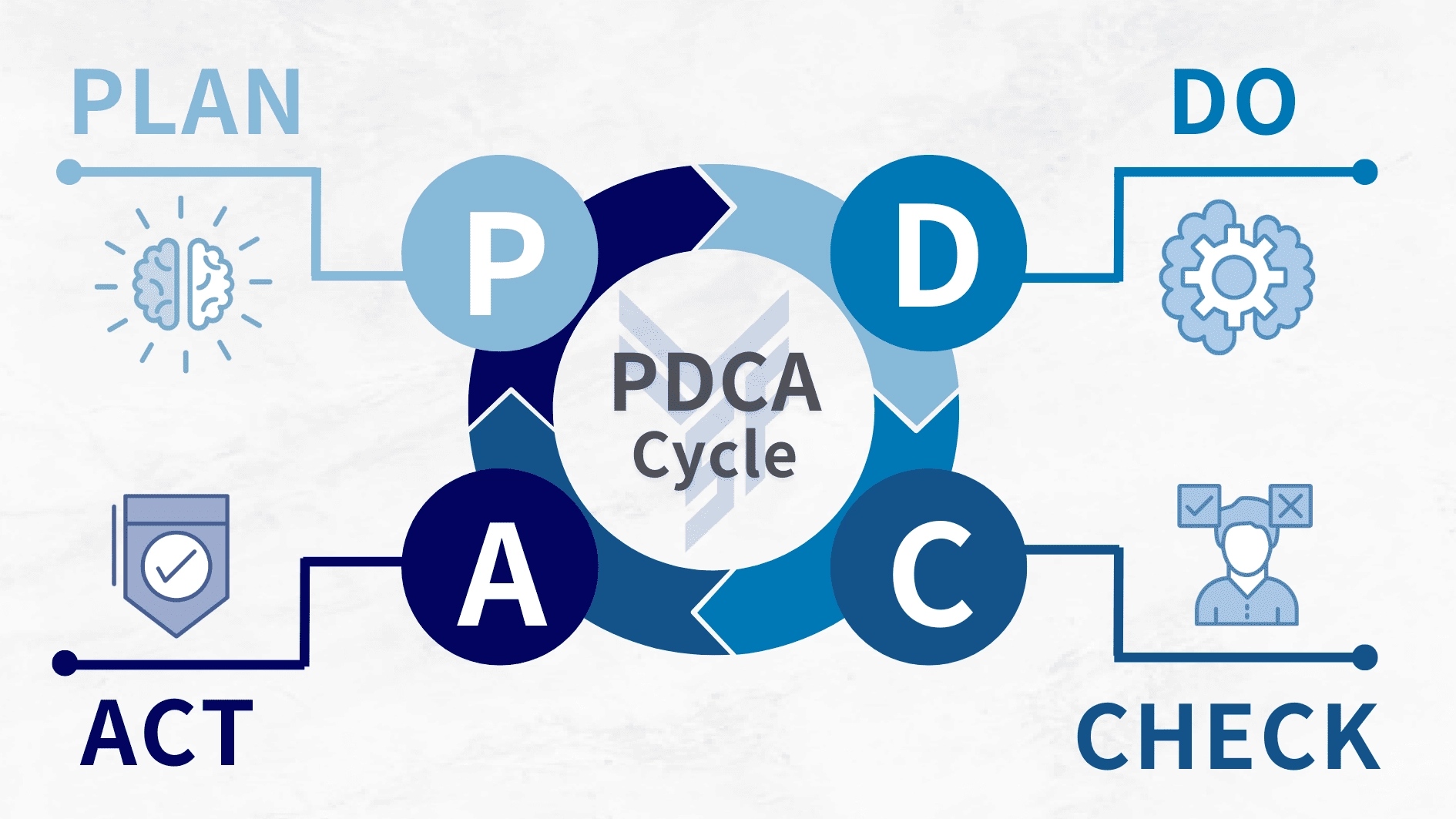
4.1 PDCA 循環的基本概念
ISO 13485 的核心價值,在於它透過 PDCA 循環讓品質管理自然融入營運流程。PDCA 起源於戴明博士的管理哲學,重點在於系統化、數據化與持續性,使企業能夠在不同階段持續觀察、調整並改善品質表現。
4.1.1 計劃 (Plan)
在計劃階段,企業需依照法規與客戶需求制定品質目標、設計製程標準並建立文件控管架構。內容通常包含產品規格、驗收標準、製程參數、控制範圍、檢驗方法與文件管理流程。以骨科植入物為例,這可能包括 Ti-6Al-4V 鈦合金的加工參數、表面處理規格與尺寸公差設定等。
4.1.2 執行 (Do)
執行階段的重點在於確保標準作業程序能被完整落實。這包含生產依照 SOP 進行、設備維護與校正、製程與檢驗數據記錄以及生產環境維持在可控狀態。實務上,製造商需建立明確的作業指導書,並確保每位操作人員通過訓練與考核。
4.1.3 檢查 (Check)
檢查階段需透過監測與稽核確認製程與產品是否符合要求。內容包括製程中的品質管控、最終產品檢驗、內部稽核與管理審查。透過數據分析,製造商能及早發現製程偏移、設備異常或人為失誤,避免問題擴大。
4.1.4 行動 (Act)
行動階段著重於根本原因分析與改善措施。當出現偏差或不符合時,需執行矯正措施與預防措施,並視需要更新文件與標準流程,使整體品質系統在每次循環後更加成熟。
4.2 PDCA 循環在醫材製造的實踐案例
以脊椎固定螺釘的生產流程為例,PDCA 循環能這樣運作。
計劃階段:制定螺紋加工的刀具路徑、切削參數與冷卻液設定,同時設定螺距公差,例如 ±0.02 mm。
執行階段:按照這些參數進行加工,並記錄每批次的加工時間、刀具磨耗情況與表面粗糙度數據。
檢查階段:使用 2.5D 影像量測儀檢測螺距,並在第十五批次開始觀察到精度偏移的趨勢。
行動階段:分析後發現原因來自刀具磨耗,將刀具更換週期由二十批次調整為十五批次,偏差問題立即獲得改善,並同步更新作業標準。
這個案例反映出 PDCA 循環的核心精神並非「事後補救」,而是在製程監控與數據分析的支持下,提前預判問題並採取預防措施。這也是 ISO 13485 強調的預防性品質管理模式。
5. 製造商面臨的挑戰與解決方案
在導入 ISO 13485 的過程中,醫療器材製造商需要面對多項組織與技術層面的挑戰。透過適當的策略與工具,這些難題都能轉化為提升品質能力與競爭力的契機。
5.1 文件管理的複雜度
ISO 13485 要求製造商建立完整的文件管理系統,內容涵蓋品質手冊、程序文件、作業指導書與各類記錄表單。對許多中小型企業而言,這是一項十分繁重的任務。
常見挑戰包括文件數量龐大、版本控管不易、跨部門協作效率低與更新過程容易出錯。有效的做法是導入電子化的文件管理系統,建立集中式的文件庫,並透過權限設定與版本控制提升準確性。同時指定專責的文件管理者,定期審查文件有效性,若搭配雲端協作平台,更能提高跨部門溝通速度與透明度。
5.2 製程驗證的資源投入
關鍵製程必須透過 IQ、OQ、PQ 驗證,證明製程能穩定地生產符合規格的產品。這對企業而言需要投入大量時間、人力與統計分析能力。
挑戰常見於驗證程序複雜、可能干擾正常生產、內部缺乏專業人才等。可行的解決方式是建立標準化的驗證計畫模板,從高風險製程優先著手,逐步擴及其他流程。必要時可尋求外部顧問協助,加速驗證進度,同時培育內部人員以建立長期的驗證能力。
5.3 供應鏈管理的挑戰
醫療器材製造涉及材料、精密零件與外包服務等多項供應來源,上游品質管理的穩定性直接影響產品的最終遵循性。
供應商品質差異大、溝通成本高與資訊透明度不足,是企業常遇到的問題。落實供應鏈管理的方式包括建立合格供應商清單、定期稽核供應商並評估其績效,對關鍵供應商要求提供品質證明文件,必要時安排駐廠檢驗。若能建立供應商分級制度,針對不同風險層級設定差異化管控措施,供應鏈的穩定度將能大幅提升。

5.4 人員訓練與品質文化建立
ISO 13485 的有效運作最終取決於人員的執行力與品質意識,因此組織文化與訓練制度扮演關鍵角色。
常見挑戰包括員工對 ISO 要求不熟悉、流程執行流於形式、缺乏改善動力等。企業可建立系統性的訓練計畫,包括品質管理基礎、作業技能訓練與品質文化導入。透過內部稽核發現問題並確保改善落實在日常作業中,搭配績效制度與改善提案獎勵,能有效提升員工參與度與品質意識。
5.5 持續改善的動力維持
取得 ISO 13485 認證只是起點,如何讓品質系統持續運作並不斷改善,是企業面臨的長期挑戰。
研發與生產團隊若在通過認證後產生鬆懈,品質系統容易逐漸僵化或變成形式上的稽核工作。建立年度品質目標與 KPI,定期進行管理審查,能確保系統持續運作。若能鼓勵員工提出改善建議、參與外部產業交流活動並將品質與業務目標連結,品質管理將不再是負擔,而是企業競爭力的重要來源。
6.元信豐的品質管理實踐
自 2014 年導入 ISO 13485 並正式轉型為醫療器材製造商以來,元信豐持續以國際標準為基礎,打造穩定、可追溯且能支援全球市場的品質管理系統。對我們而言,品質管理不只是為了通過認證,而是確保產品安全、降低風險並協助客戶在各國市場順利上市的核心能力。

6.1 持續改善的動力維持全流程品質控管
元信豐在產品全生命週期中建立完整的品質控管流程,從設計對接、原料進料檢驗、製程參數設定、製程監控、品質檢驗到包裝與售後服務,每個階段都有明確的作業標準與記錄要求。我們使用 2.5D 影像量測儀、粗度儀與各式精密檢查設備,確保每一件骨科植入物都符合尺寸精度與表面品質標準。
6.2 風險管理機制
針對各項產品線,我們依照 ISO 14971 進行系統化的風險評估,辨識可能的失效模式並在製程中建立控制措施。以 Ti-6Al-4V 鈦合金加工為例,我們透過自家開發的冷卻技術降低加工時的溫度波動,避免材料性能因過熱而劣化,確保植入物在臨床使用中維持穩定的機械性能與生物相容性。
6.3 文件追溯系統
我們建立完整的產品履歷紀錄,包含原料批號、加工設備、製程參數、操作人員、檢驗結果與包裝出貨資料,讓每一件產品都能「從原料到成品」完整追溯。這讓我們能在客戶稽核或主管機關要求提供文件時,即時提供完整資料,也大幅降低供應鏈風險。
6.4 持續改善文化
元信豐以 PDCA 循環為核心進行品質改善,定期進行內部稽核與管理審查,以數據為基礎檢視品質系統的運作狀況。我們鼓勵員工提出改善提案,並以實際成果作為績效回饋,使品質文化能在日常工作中自然落地,持續提升製程效率並降低不良率。
作為具備完整實績的一站式醫療器材代工夥伴,元信豐能協助客戶從樣品試製、製程導入、技術文件準備到面對第三方稽核的全套流程。我們的品質管理能力已成功支援歐美日等高規市場,協助客戶以更快速度、更高穩定度進入國際醫材供應鏈。
7.常見問題(FAQ):ISO 13485認證實務問答
1. ISO 13485 是什麼?為什麼醫療器材產業需要它?
ISO 13485 是專為醫療器材產業制定的品質管理系統標準,用來確保產品能在整個生命週期中保持安全、有效及符合法規要求。它涵蓋設計、開發、生產、包裝、滅菌、倉儲、配送與售後活動,是全球多國監管機構共同依循的重要框架。企業導入 ISO 13485 不僅能強化品質一致性,也能提升可追溯性、風險管理能力與文件透明度。對醫材製造商而言,ISO 13485 是取得國際客戶信任與進入全球供應鏈的核心門檻。透過完善的系統管理,企業能降低不良率、縮短客戶審查時間並提升市場競爭力。
2.ISO 13485 與 ISO 9001 有何差異?哪一個對醫療產業更重要?
ISO 9001 是適用於各行業的一般品質管理系統,而 ISO 13485 則專注於醫療器材領域,強調法規遵循、產品安全與風險管理。最明顯的差異在於其管理思維,ISO 13485 採「法規優先」,要求企業依照各國監管要求建立流程,例如追溯性管理、風險控制、設計驗證與製程驗證等。ISO 9001 追求客戶滿意度,而 ISO 13485 更強調產品在臨床使用時的安全性。對醫療器材製造商而言,ISO 13485 是不可或缺的核心標準,能協助企業符合全球法規要求,也能提升與國際品牌合作的機會。
3.有了 ISO 13485 是否就能把產品賣到美國、歐洲或日本?
ISO 13485 是國際採認度最高的醫療器材品質管理系統,但它本身不是產品上市許可,也不能取代各國法規核可。例如,美國市場仍需取得 FDA 510k 或 PMA,歐盟需完成 CE 技術文件審查,日本則需符合 PMDA 的審查要求。ISO 13485 的作用在於確保企業具備符合法規的品質管理能力,能減少審查過程中的問題,也讓技術文件更容易通過評估。因此,ISO 13485 是「基礎門檻」,真正上市仍需依市場取得相對應的產品認證。
4. ISO 13485 的認證需要多久?取得後還要做什麼?
導入 ISO 13485 時間因企業規模與制度成熟度而異,一般需六個月到一年。過程包含流程建立、文件編寫、風險管理、製程驗證、訓練以及內部稽核。取得認證後,企業仍需每年接受監督稽核並持續更新文件、改善流程、維持追溯性與風險管理。ISO 13485 的精神是 PDCA 持續改善,因此認證不是終點,而是要求企業不斷維護品質系統、降低市場風險並回應法規更新。穩定的長期維運,才能讓 ISO 系統真正發揮價值。
5.如何確認 ISO 13485 證書是否為真?如何避免合作風險?
驗證 ISO 13485 證書真偽的方式包括確認證書上的認證機構是否為國際認可的第三方,例如 SGS、BSI、DNV、TUV 等。可以直接到發證機構網站輸入證書編號驗證有效性,並檢查證書範圍是否涵蓋相關的製造活動與產品類別。若製造商願意提供外部稽核報告、不符合項與改善紀錄,也代表其品質系統較為成熟。最可靠的方法是進行現場稽核,確認企業日常作業與文件記載是否一致。透明度越高的製造商,通常品質系統越值得信賴。
8.結論:ISO 13485不只是認證,更是競爭力
在醫療器材產業中,ISO 13485 已從「選配」變成「標配」。它不僅是進入國際市場的基本門檻,更是建立品質信譽與提升競爭力的關鍵基礎。對品牌商而言,選擇具備 ISO 13485 認證的 OEM 夥伴,可以有效降低供應鏈風險,確保產品品質與批次間的一致性。對製造商而言,雖然導入與維護 ISO 13485 需要投入時間與資源,但長期帶來的效益,在風險控管、市場信任與營運效率上,往往遠高於成本。
從系統化的製程管理、完整的紀錄與追溯,到持續改善與風險管理機制,ISO 13485 提供的不只是一套標準,更是一種管理哲學。它協助製造商從「事後品檢」走向「事前預防」,從「經驗導向」走向「數據導向」,從「依賴個人」走向「依循系統」。在高風險、高監管要求的醫療器材領域,這樣的轉變是企業得以長期穩定成長的必要條件。
元信豐深耕骨科醫材精密加工逾三十年,自 2014 年導入 ISO 13485 以來,持續以國際標準為基礎,建立與歐美日市場接軌的品質管理系統。我們不僅專注於提供符合國際規範的骨科植入物與相關醫療器材產品,也協助客戶優化製程、提升品質效能並強化法規應對與全球市場佈局。
如果您正在尋找值得信賴的醫療器材 OEM 合作夥伴,歡迎與元信豐聯繫。我們將以扎實的加工經驗、完整的品質系統與專業團隊,協助您在全球醫療器材市場中穩健發展,讓產品在安全、品質與速度之間取得最佳平衡。
若您有開發專案或代工需求,歡迎填寫聯絡表單或來信至 sales@ysfbone.com,我們將有專人於24小時內回覆您。
9.聲明
本文內容僅供醫療產業與專業人士參考之用,部分資訊來自公開資料或專家觀點整理,難免有所遺漏或需進一步補充,誠摯歡迎交流與指正。
提醒您:任何醫療診斷與治療決策,仍應以合格臨床醫師之專業評估與建議為唯一依據,患者切勿僅憑本文資訊自行做出醫療相關決定。
10.參考資料
ISO 13485標準與相關資源
International Organization for Standardization. (2016). ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes.
https://www.iso.org/standard/59752.html
International Organization for Standardization. (n.d.). ISO 13485 — Medical devices.
https://www.iso.org/iso-13485-medical-devices.html
FDA相關資源
U.S. Food and Drug Administration. (2024, February 2). Medical devices; Quality system regulation amendments [Final rule]. Federal Register, 89(22), 7496-7650. https://www.federalregister.gov/documents/2024/02/02/2024-01709/medical-devices-quality-system-regulation-amendments
U.S. Food and Drug Administration. (2024). Quality Management System Regulation: Final rule amending the Quality System Regulation – Frequently asked questions.
https://www.fda.gov/medical-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp/quality-management-system-regulation-final-rule-amending-quality-system-regulation-frequently-asked
U.S. Food and Drug Administration. (2013, September 24). Unique Device Identification System [Final rule]. Federal Register, 78(185), 58785-58828. https://www.federalregister.gov/documents/2013/09/24/2013-23059/unique-device-identification-system
U.S. Food and Drug Administration. (n.d.-b). About Manufacturer and User Facility Device Experience (MAUDE) database.
https://www.fda.gov/medical-devices/mandatory-reporting-requirements-manufacturers-importers-and-device-user-facilities/about-manufacturer-and-user-facility-device-experience-maude-database
U.S. Food and Drug Administration. (n.d.-c). Federal Food, Drug, and Cosmetic Act (FD&C Act).
https://www.fda.gov/regulatory-information/laws-enforced-fda/federal-food-drug-and-cosmetic-act-fdc-act
U.S. Food and Drug Administration. (2011). Process validation: General principles and practices [Guidance for industry].
https://www.fda.gov/media/71021/download
Legal Information Institute, Cornell Law School. (n.d.-a). 21 CFR Part 820 - Quality System Regulation.
https://www.law.cornell.edu/cfr/text/21/part-820
Covington & Burling LLP. (2024, February). FDA Finalizes Rule Incorporating ISO 13485 into New Quality Management System Regulation (QMSR).
https://www.cov.com/en/news-and-insights/insights/2024/02/fda-finalizes-rule-incorporating-iso-13485-into-new-quality-management-system-regulation-qmsr
Morgan Lewis. (2024, October 10). February 2, 2026 Is Quickly Approaching—Are You QMSR Ready? https://www.morganlewis.com/pubs/2024/10/february-2-2026-is-quickly-approaching-are-you-qmsr-ready
Greenlight Guru. (2024, February 2). FDA QMSR: QSR, ISO 13485 & Harmonization Explained.
https://www.greenlight.guru/blog/qmsr-quality-management-system-regulation
Emergo by UL. (n.d.-a). US FDA Incorporates ISO 13485 Within Its QMSR Final Rule.
https://www.emergobyul.com/news/us-fda-incorporates-iso-13485-within-its-qmsr-final-rule
The FDA Group. (2024, March 19). FDA's Quality Management System Regulation (QMSR): A Quick-Guide. https://www.thefdagroup.com/blog/qmsr-quality-management-system-regulation
Qualityze. (n.d.). Impact of the FDA's QMSR Final Rule on Global Markets.
https://www.qualityze.com/blogs/fda-qmsr-final-rule-on-global-markets
Arnold & Porter. (2024, February). FDA Issues Final Rule to More Closely Align FDA Medical Device Quality System Requirements With International Consensus Standard ISO 13485. https://www.arnoldporter.com/en/perspectives/advisories/2024/02/fda-final-rule-medical-device-requirements-and-iso-13485
Stanton, T. (2024, October 31). FDA facing increased scrutiny over medical device recalls. MD+DI.
https://www.mddionline.com/regulatory-quality/fda-facing-increased-scrutiny-over-medical-device-recalls
歐盟MDR/IVDR相關資源
European Union. (2017a). Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices [Medical Device Regulation]. Official Journal of the European Union, L 117.
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
European Union. (2017b). Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices [PDF].
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745
European Union. (2017c). Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. Official Journal of the European Union, L 117.
https://eur-lex.europa.eu/eli/reg/2017/746/oj
European Commission, Single Market Economy. (n.d.). Harmonised standards for medical devices.
https://single-market-economy.ec.europa.eu/single-market/goods/european-standards/harmonised-standards/medical-devices_en
Johner Institute. (2025, February 4). EU Medical Device Regulation MDR: Everything you need to know.
https://blog.johner-institute.com/regulatory-affairs/medical-device-regulation-mdr/
Advisera. (2024, November 19). MDR compliance - How to achieve it, and can ISO 13485 help? https://advisera.com/13485academy/blog/2020/03/09/how-can-iso-13485-help-with-mdr-compliance/
Decomplix. (2025, July 17). Understanding Harmonized Standards for medical devices and IVDs.
https://decomplix.com/understanding-harmonized-standards-medical-devices-ivds/
Kobridge Consulting. (2022, June 1). EU MDR harmonized standards - first set released.
https://kobridgeconsulting.com/eu-mdr-harmonized-standards/
Medical Device Regulation. (2023, March 3). MDR harmonized.
https://www.medical-device-regulation.eu/2023/03/03/mdr-harmonized/
Sobel Consult. (2025, June 1). MDR 2017/745 vs ISO 13485: Decoding Quality Management Requirements.
https://sobelconsult.com/mdr-2017-745-vs-iso-13485-decoding-quality-management-requirements/
EN ISO 13485:2016+A11:2021及附錄ZA/ZB
Qarad. (2024, October 25). EN ISO 13485:2016+A11:2021 published.
https://qarad.com/news/en-iso-134852016a112021-published/
Critical Catalyst. (n.d.). EN ISO 13485:2016 – Amendment 11 has been published!
https://criticalcatalyst.com/en-iso-134852016-amendment-11-has-been-published/
BSI Group. (n.d.-a). Amendment to EN ISO 13485:2016 published. https://compliancenavigator.bsigroup.com/en/medicaldeviceblog/european-amendment-published-for-medical-devices-quality-management-system-standard/
BSI Group. (n.d.-b). Further amendment to lists of harmonised standards with presumption of conformity to European medical devices regulations. https://compliancenavigator.bsigroup.com/en/medicaldeviceblog/further-amendment-to-lists-of-harmonised-standards/
Colabon. (2025, May 12). SN EN ISO 13485 A11 2021. https://www.colabon.com/en/post/sn-en-iso-13485
Qserve Group. (2021, September 19). EN ISO 13485:2016+A11:2021 - It has been published!
https://qservegroup.com/blog/en/en-iso-134852016a112021-it-has-been-published
Qualitiso. (2021, September 29). EN ISO 13485/A11: Annex ZA and ZB for regulations 2017/745 and 2017/746.
https://www.qualitiso.com/en/en-iso-13485-a11/
MedEnvoy Global. (2025, May 5). ISO 13485 Medical Devices: Global Regulatory Compliance.
https://medenvoyglobal.com/blog/iso-13485-medical-devices-global-regulatory-compliance/
UDI與EUDAMED
Johner Institute. (2025, February 19). Unique Device Identification (UDI) in the EU.
https://blog.johner-institute.com/regulatory-affairs/unique-device-identification-udi/
GS1. (n.d.). Unique Device Identification (UDI) - Healthcare. https://www.gs1.org/industries/healthcare/udi
Greenlight Guru. (n.d.-a). Explaining UDI Requirements for GUDID and EUDAMED.
https://www.greenlight.guru/blog/udi-gudid-eudamed
European Commission. (n.d.-a). UDI/Device registration - Public Health.
https://health.ec.europa.eu/medical-devices-eudamed/udidevice-registration_en
European Commission. (n.d.-b). Legacy device management [PDF]. https://health.ec.europa.eu/document/download/4699ad8d-bf2c-4303-bd25-10e26770c8b1_enfilename=legacy_dvc_management_en.pdf
DDI Smart. (2024, May 31). UDI & EUDAMED Explained under EU MDR.
https://www.ddismart.com/blog/udi-eudamed-explained-under-eu-mdr/
Easy Medical Device. (2024, October 4). UDI Beginners Guide: Unique Device Identification (EU MDR and IVDR).
https://easymedicaldevice.com/udi/
Kalypso. (2024, January 25). Leading Practices for EUDAMED and Basic UDI.
https://kalypso.com/viewpoints/entry/leading-practices-for-eudamed-and-basic-udi
Open Regulatory. (n.d.). What is a MDR UDI? Unraveling the Importance of Unique Device Identification.
https://openregulatory.com/questions/what-is-a-mdr-udi
Rimsys. (n.d.-a). The ultimate guide to the EU MDR/IVDR UDI. https://www.rimsys.io/blog/the-ultimate-guide-to-the-eu-mdr-ivdr-udi
MedTech Europe. (2017, August). European Unique Device Identification Database (EUDID) [Background paper]. https://www.medtecheurope.org/wp-content/uploads/2017/08/2014_MTE_Background-Paper_EU-UDI-Database.pdf
Post-Market Surveillance與Vigilance System
Mantra Systems. (2025, July 22). Post-Market Surveillance (PMS): Understanding PMCF & Vigilance under the EU MDR. https://mantrasystems.com/articles/pms-understanding-pmcf-vigilance-under-eu-mdr
Mantra Systems. (2025, October 13). Vigilance and Post-Market Surveillance (PMS).
https://mantrasystems.com/eu-mdr-compliance/vigilance
Mantra Systems. (2025, March 31). Post-Market Surveillance (PMS) of medical devices.
https://mantrasystems.com/eu-mdr-compliance/post-market-surveillance-pms
European Commission. (n.d.-c). Market surveillance and vigilance - Public Health.
https://health.ec.europa.eu/medical-devices-sector/directives/market-surveillance-and-vigilance_en
Rimsys. (n.d.-b). Post-market surveillance for medical devices in the European Union.
https://www.rimsys.io/blog/post-market-surveillance-for-medical-devices-in-the-european-union
Greenlight Guru. (n.d.-b). Postmarket Surveillance of Medical Devices: Ultimate Guide.
https://www.greenlight.guru/blog/postmarket-surveillance
VDE. (n.d.). Post-Market Surveillance (PMS) and Vigilance of Medical Devices according to MDR.
https://www.vde.com/topics-en/health/consulting/pms-and-vigilance-of-medical-devices-according-to-mdr
EUMDR. (n.d.). Post Market Surveillance System – Regulation (EU) 2017/745 (EU MDR).
https://eumdr.com/post-market-surveillance-system/
RegDesk. (2025, March 20). Understanding the Post-Market Surveillance Requirements Under the EU MDR. https://www.regdesk.co/understanding-the-post-market-surveillance-requirements-under-the-eu-mdr/
台灣TFDA相關資源
衛生福利部. (2021年4月14日). 醫療器材品質管理系統查核準則 [公告]. 全國法規資料庫.
https://law.moj.gov.tw/LawClass/LawAll.aspx?pcode=L0030116
衛生福利部食品藥物管理署. (n.d.). 醫療器材製造業者品質管理系統(QMS)申請.
https://www.fda.gov.tw/Tc/siteContent.aspx?sid=11584
財團法人塑膠工業技術發展中心. (n.d.). 中華民國|國產醫療器材QMS登錄輔導.
https://www.pidc.org.tw/materials.php?id=830
Pacific Bridge Medical. (2025, January 28). Medical Device Registration in Taiwan.
https://www.pacificbridgemedical.com/regulatory-services/medical-device/product-registration/taiwan/
Asia Actual. (2025, May 20). Taiwan Medical Device Registration and Approval.
https://asiaactual.com/taiwan/medical-device-registration/
Pure Global. (n.d.). FDA Taiwan Medical Device Registration. https://www.pureglobal.com/markets/taiwan
Qserve Group. (n.d.). Taiwan Medical Device Regulations. https://qservegroup.com/en/taiwan-medical-device-regulations
日本PMDA相關資源
Pharmaceuticals and Medical Devices Agency. (2021, March 26). Revision of Japanese Medical Device QMS requirements. https://www.pmda.go.jp/english/review-services/regulatory-info/0004.html
Emergo by UL. (n.d.-b). Quality Management System Compliance with Japan Ordinance 169.
https://www.emergobyul.com/services/quality-management-system-compliance-japan-ordinance-169
Emergo by UL. (2024, October). Japan MHLW Ordinance 169 and Medical Device and IVD QMS requirements [White paper]. https://www.emergobyul.com/sites/default/files/2024-10/Japan-MHLW-Ordinance-IVD-QMS-requirements-Whitepaper.pdf
BSI Canada. (n.d.). MDSAP Revision of MHLW MO169, Japan medical device QMS requirements.
https://www.bsigroup.com/en-CA/Medical-Devices/news-centre/Enews/2022-news/mdsap-revision-of-mhlw-mo169/
Qtec Group. (2025, July 25). Approval of Medical Devices in Japan.
https://www.qtec-group.com/en/zulassung-von-medizinprodukten-in-japan/
Biosector Ltd. (2024, February 28). ISO 13485 in Japan - the Global Standard for Medical Device Quality.
https://biosector.jp/iso_13485_in_japan/
Evershine. (2023, July 17). Japan DMAH Practice QA. https://tyo.evershinecpa.com/japan-dmah-practice-qa
PMDA. (n.d.). Regulations for QMS and SaMD in Japan [PDF]. https://www.pmda.go.jp/files/000269712.pdf
品質管理工具與方法
American Society for Quality. (n.d.). What is Failure Mode and Effects Analysis (FMEA)?
https://asq.org/quality-resources/fmea
PTC. (2025, September 18). ISO 13485 for Medical Devices QMS: The Complete Guide.
https://www.ptc.com/en/blogs/medtech/iso-13485-qms-medical-device
Greenlight Guru. (n.d.-c). ISO 13485 for Medical Devices QMS [Complete Guide].
https://www.greenlight.guru/blog/iso-13485-qms-medical-device
DNV. (n.d.). ISO 13485 – Medical devices quality management. https://www.dnv.us/services/iso-13485-medical-devices-quality-management-235016/
NSF. (2020, February 5). ISO 13485:2016 Certification: Medical Devices QMS.
https://www.nsf.org/management-systems/quality-management/iso-13485
產業資源與研究
Peters, W., Pellerin, C., & Janney, C. (2020). Analysis: Using the FDA MAUDE and Medical Device Recall databases to design better devices. Biomedical Instrumentation & Technology, 54(3), 178-194.
https://doi.org/10.2345/0899-8205-54.3.178
© 2025 元信豐企業股份有限公司 |骨科植入物及手術器材製造專家、ISO 13485 認證醫材廠、CNC 精密加工 |引用或轉載請註明來源,版權所有。




