
摘要
在醫療器材供應鏈中,挑選可靠的 OEM 製造夥伴是影響產品品質、法規順利性與上市時程的關鍵決策。尤其是骨科植入物等高風險、高精密產品,製造端的品質管理成熟度將直接左右批次穩定性與臨床安全。本篇文章從 ISO 13485 的實務運作出發,說明如何評估代工廠的品質系統、風險管理、製程驗證、供應鏈控管與稽核配合能力,並分享元信豐累積多年的品質管理經驗,協助品牌商在選擇 OEM 夥伴時做出更精準的判斷。
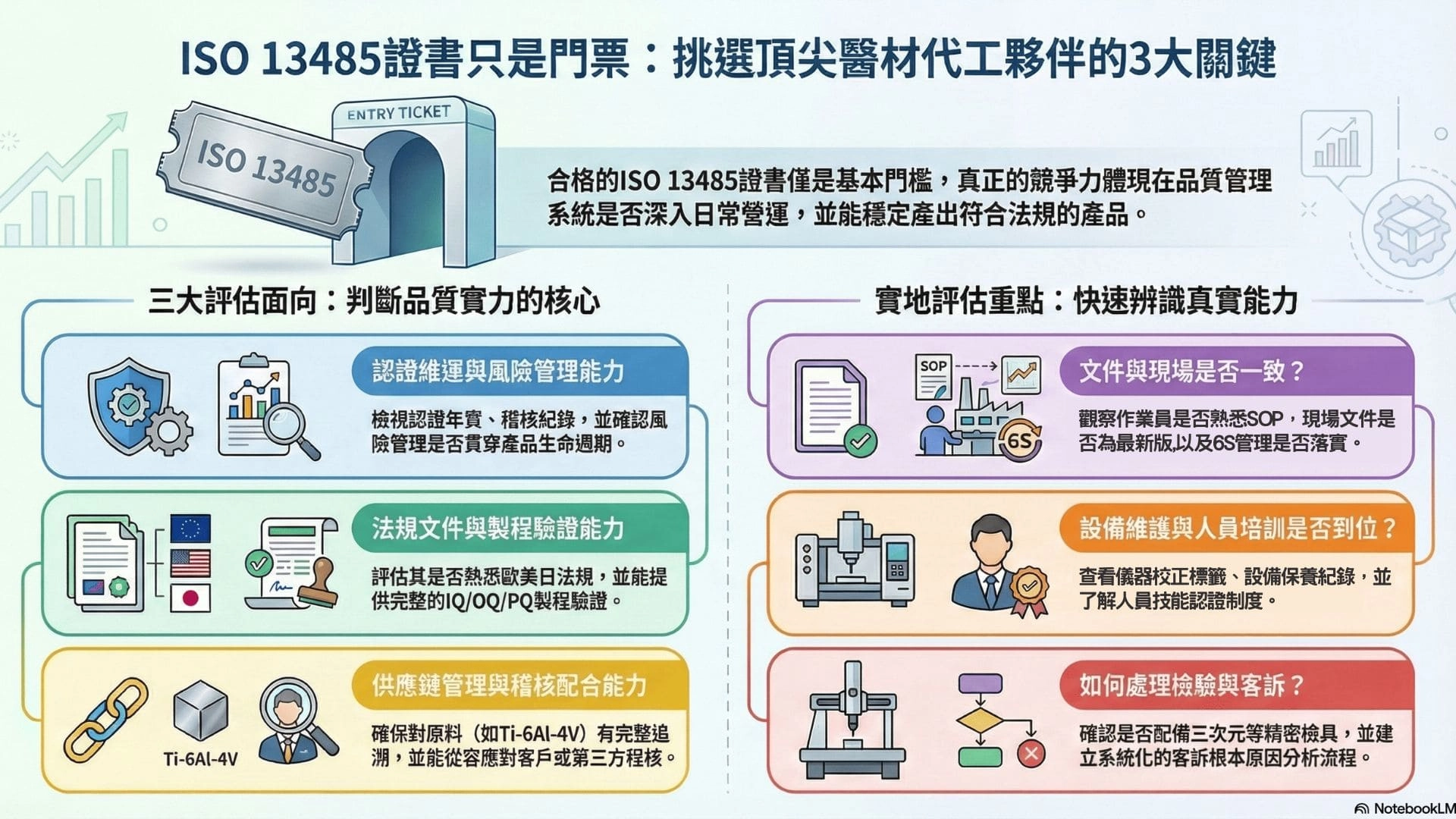
Table of Contents
1.前言:合作代工廠,首先確認品質系統是否到位
1-1 品質系統會決定合作成效
醫療器材 OEM 合作中最常見的風險,不是設備不夠好、經驗不夠多,而是品質管理系統沒有真正運作。若追溯文件不完整、風險管理未落實、系統化作業不健全,就容易在量產階段出現批次不穩定或文件延誤,甚至無法通過客戶稽核。
特別是骨科植入物等需長期留置體內的產品,任何加工中細微的變異都可能帶來臨床風險,因此評估代工廠時,品質管理能力永遠比報價更重要。
1-2 全球法規趨嚴,使合作模式從價格走向價值
醫療器材的法規要求提升後,品牌商逐漸不再以價格作為唯一指標,而是關注代工廠是否能提供穩定、可追溯、符合法規的完整製造服務。一家成熟的 OEM 工廠能在設計階段提供可製造性分析,在量產階段維持一致性,在上市後協助品質監控,為客戶帶來更高的整體合作價值。
1-3 ISO 13485 的核心意義
ISO 13485 代表製造商具備了基本的醫療器材品質管理架構,涵蓋設計、生產、檢驗、包裝、出貨等環節。取得證書僅代表通過審查,而真正能展現競爭力的是持續維運這套系統,包括日常稽核、文件管理、風險分析、數據監控與內部改善等。
「評估代工廠時,不只看證書,更要確認系統是否真的落地執行。」
2. 三大評估面向:判斷醫材代工廠品質實力的關鍵
2-1 認證維運與風險管理能力
長期穩定維持 ISO 13485 認證,意味著品質系統已內化進日常作業。評估代工夥伴時,可觀察以下項目:
認證維運狀況:
-
認證年資是否足夠
-
認證範圍是否涵蓋您的產品類別
-
是否曾因重大缺失而停證
-
最近三年外部稽核結果與改善情況
風險管理成熟度:
醫材風險管理需貫穿產品生命週期。以骨科植入物為例,需評估:
-
材料生物相容性
-
加工溫度與刀具磨耗影響
-
表面處理後可能產生的風險
-
尺寸偏差可能造成的臨床影響
-
包裝與運輸造成污染的風險
成熟的製造商會透過製程監控、統計分析與異常預警,將風險處理在製造端,而非依賴最終檢驗找問題。
2-2. 法規文件配合與製程驗證能力
醫療器材上市需要準備大量技術文件,具備豐富經驗的代工廠能夠理解不同市場的文件要求差異,主動協助客戶準備完整資料,例如美國市場要求的 Device History Record 需要完整批次追溯紀錄,歐盟 MDR 強調技術文件完整性與臨床評估,日本 PMDA 則要求詳細的Medical Device File,品牌商通常會進行現場稽核或定期查廠,專業的製造商能配合稽核安排、提前準備相關文件,並在稽核後快速回應缺失事項。
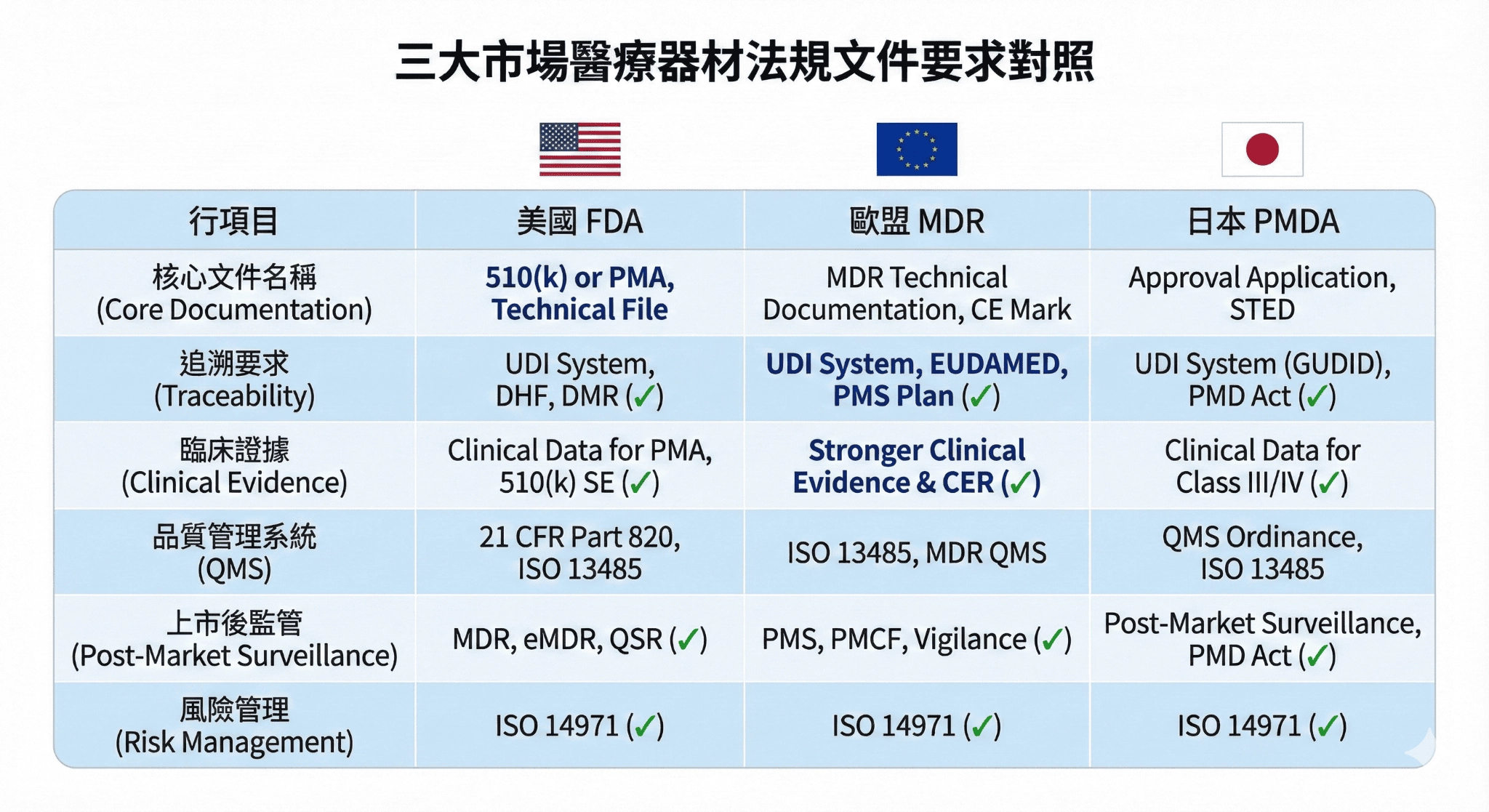
表1:美國、歐盟、日本等三大市場醫療器材法規文件要求對照(2025元信豐企業整理)
雖然多數OEM合作由客戶提供設計,但製造商仍需管理設計輸入與輸出、確認製造可行性、進行必要的設計驗證,專業代工廠會建立設計審查機制,評估可製造性、風險程度、驗證需求,維護完整的設計文件,建立變更管理流程確保任何設計調整都經過評估與驗證,關鍵製程必須透過 IQ(安裝確效)、OQ(運轉確效)、PQ(性能確效)驗證,以證明製程能穩定生產符合規格的產品。評估時可查看:
-
是否建立標準化的驗證程序
-
驗證計畫是否完整
-
是否涵蓋最壞情境
-
測試數據是否完整記錄
-
是否有定期再驗證制度
2-3 供應鏈管理與稽核配合能力
高品質的醫療器材製造仰賴穩定的供應鏈,而骨科植入物使用的 Ti-6Al-4V、316L不鏽鋼與 PEEK 等材料,都需要具備完整的材料證明與批號追溯。成熟的 OEM 製造商會以制度化方式管理供應鏈,包含供應商的資格評鑑、風險等級分類、定期查廠與績效追蹤,並建立清楚的材料管理流程,使原料從採購、入庫、檢驗到投料都能維持一致性與可追溯性。
當客戶或第三方機構安排稽核時,專業代工廠能提出完善準備,包括事先蒐集文件、安排場區動線、解釋流程與呈現佐證資料,也會在稽核後主動完成改善,使整個合作流程更透明、更具可控性。
3. 證書背後:品質管理的真實價值
3-1 文件以外的實際運作
ISO 13485 證書象徵廠商具備基本品質制度,但是否真正落實到現場,仍需觀察日常運作。品質系統若真正內化,現場作業員能清楚說明標準作業流程,文件也能在現場即時取得與更新。內部稽核會規律執行,異常事件能被迅速記錄並追蹤,製程偏差也能透過數據監控在早期被識別。這些環節都直接影響製程穩定度與產線的一致性,也使客戶在專案開發與法規審查階段更加順利。

3-2 以數據推動改善的組織文化
醫療器材製造講求可控性與可預測性,因此以數據作為管理依據十分重要。具備成熟品質文化的工廠,習慣透過不良率趨勢、製程能力數據、設備狀態、客訴統計與品質成本等資訊來監控整體表現,並在數據出現異常時迅速採取行動。改善通常由跨部門共同執行,而第一線員工也是改善來源的重要角色,因為他們最接近問題現場,能直接提出實務性的建議。透過這樣的循環,品質系統能持續成長,生產變異自然逐年下降。
4.實地評估重點:從現場快速辨識代工廠的真實能力
4-1 文件系統與現場管理
實際參訪代工廠時,最能反映管理能力的是文件是否與現場一致,以及生產環境是否維持整齊與明確規範。成熟工廠多半使用電子化文件管理系統,讓每位作業人員都能即時看到最新版本的作業指導書與檢驗要求。生產區的動線、物料擺放與標示清晰度,也能直接反映工廠對製程穩定度的重視程度。6S 包含:整理(Seiri)、整頓(Seiton)、清掃(Seiso)、清潔(Seiketsu)、習慣(Shitsuke)、安全(Safety),良好的 6S 管理讓操作環境更有秩序,並降低人為錯誤、設備碰撞與交叉污染的風險。

4-2. 設備維護與人員培訓
精密加工設備必須維持最佳狀態才能生產高品質醫材,因此設備履歷、保養計畫、故障紀錄與校正報告,都是評估工廠是否具備長期穩定能力的重要依據。若量測儀器的校正貼紙都是最新、儀器狀況清楚可追溯,代表工廠對品質控制有系統性的安排。同時,人員培訓制度也能看出工廠對風險的理解程度。具備完善訓練矩陣與技能認證制度的工廠,會讓每位員工在進線前經過明確的訓練流程,也會提供定期複訓,使能力維持在可控範圍。

4-3. 檢驗能力與客訴處理
骨科植入物需要進行尺寸檢驗、表面品質檢驗、機械性能測試等多項檢驗,工廠必需配備相應的檢驗設備,包括三次元量測儀、2.5D 投影機、表面粗度儀、光學檢驗設備等,更重要的是培養專業的品質檢驗人員,確保檢驗方法正確、判定標準一致、紀錄完整清楚。
此外,也需建立完整的客訴處理機制,包括客訴接收、分類、調查、改善、回覆、追蹤等流程,他們視客訴為改善機會,會進行根本原因分析並制定預防措施,定期分析客訴趨勢以發現系統性問題。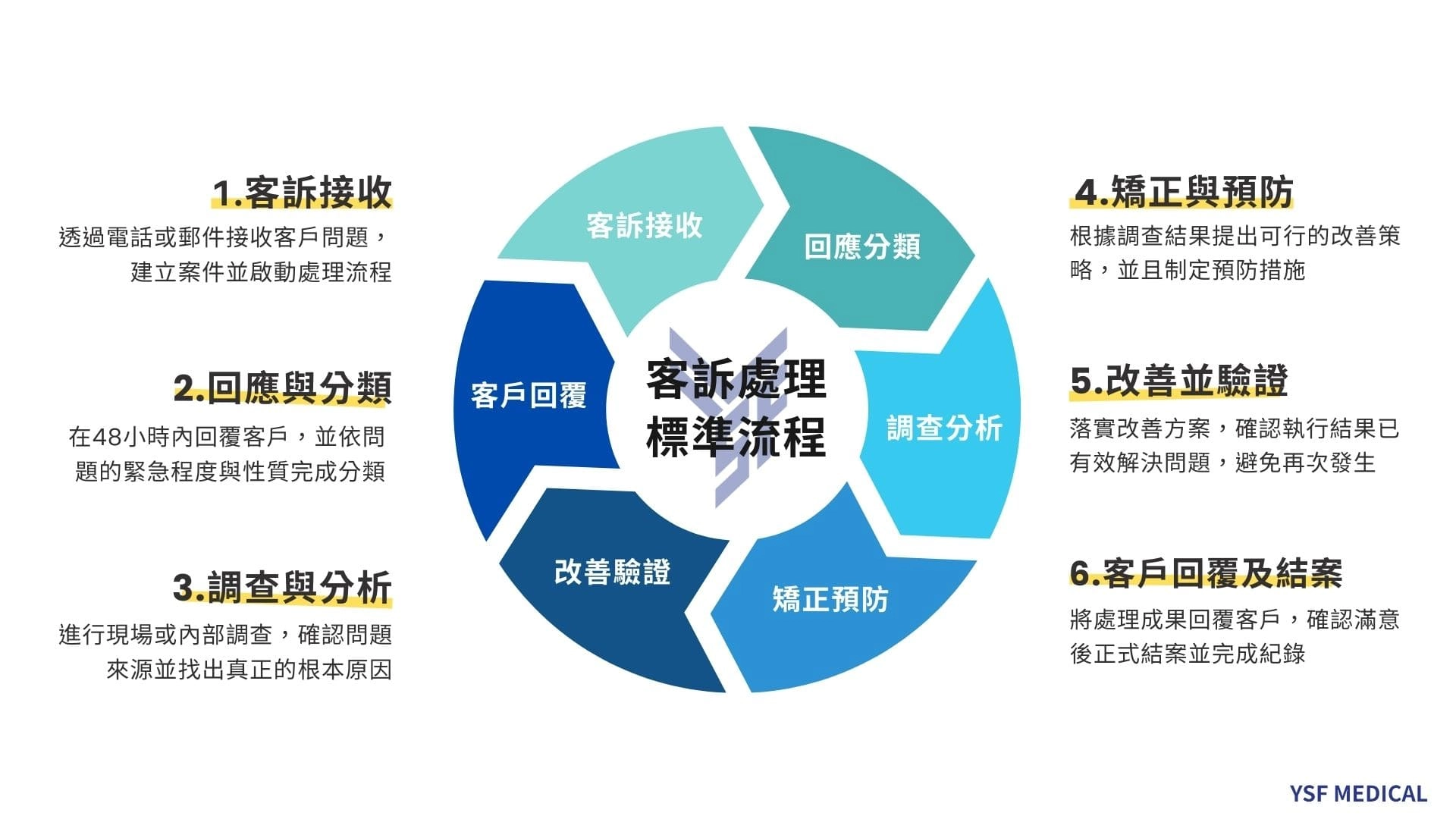
5. 元信豐的品質管理實踐
5-1. 完整的品質管理體系
元信豐自 2014 年轉型為醫療器材製造商後,依據 ISO 13485 建立完整品質管理體系。我們累積了來自不同市場類型客戶的查廠與稽核經驗,並持續根據稽核結果與產品特性改善系統。
我們建立從設計對接、原料管理、製程驗證到成品檢驗與包裝文件的全流程控管。關鍵製程皆已通過 IQ、OQ、PQ 驗證,並維護標準化作業程序。
同時依據 ISO 14971 持續執行風險分析,建立材料、工具、操作與環境面向的控制措施,使製程穩定度持續提升,以Ti-6Al-4V鈦合金加工為例,我們評估加工溫度過高、刀具磨耗、切削液污染、人為失誤等風險,並建立相應的控制措施,包括自主開發的冷卻技術、定期更換刀具並監控切削狀態、嚴格的切削液管理、標準化作業程序與技能認證。
5-2. 持續改善與量化成果
我們以PDCA循環為核心進行品質改善,定期進行內部稽核與管理審查,鼓勵員工提出改善提案並將實際成效作為績效回饋,推動的改善專案包括優化冷卻系統降低加工溫度變異、導入自動化檢驗設備提升效率、建立供應商評鑑制度強化供應鏈管理、升級文件管理系統提升追溯效率。
近年品質指標如下:
-
製程良率高於 95%
-
準時交期達成率 97%
-
客戶抱怨件數低於5件
-
近三十次客戶稽核皆無重大缺失
這些成果反映品質系統已內化至日常作業,而非僅維持證書標準。
5-3. 一站式服務能力
元信豐提供從樣品試製、製程導入、文件協助到量產製造與稽核配合的完整服務。我們的團隊能協助客戶處理可製造性分析、風險管理、製程驗證與追溯文件準備,使產品以更快速度、更高穩定度進入國際醫材供應鏈。
6.常見問題:挑選代工夥伴實務問答
Q1:第一次與醫療器材代工廠接觸時,應該確認哪些最重要的資訊?
初次接觸時,建議以品質管理系統與醫材製造經驗作為核心判斷基準。可先詢問代工廠的 ISO 13485 認證取得時間、認證範圍、近三至五年的外部稽核紀錄與不符合項改善情況,這些資訊能反映工廠是否具備長期維運品質系統的能力。
接著,可以了解公司是否擁有專責品質部門、具體人員配置與專業背景,並詢問是否曾代工相似產品,例如骨科植入物、手術器械或高精密醫療器材。
此外,建議進一步詢問代工廠如何處理批次異常、如何追溯材料來源與製程紀錄,以及是否有標準化 CAPA 機制,能快速判斷工廠是否具備醫材 OEM 所需的專業能力。
Q2:若多家代工廠都擁有 ISO 13485 認證,該如何比較品質管理的差異?
ISO 13485 是醫療器材 OEM 產業的基本門檻,但真正的差異在於品質管理系統是否有落實到日常生產。比較時可觀察認證年資長短、適用範圍是否涵蓋您的產品類別,以及代工廠是否能提供稽核報告與改善紀錄,這些資料展現的是品質系統的成熟度。除了證書本身,更重要的是要求代工廠提供客觀的品質績效數據,例如製程不良率、交期達成率、材料追溯完成度、量測能力指標、客訴回應時效等。
若兩家工廠的設備相似,通常差異會出現在品質文化。例如是否定期執行內部稽核、是否有跨部門改善機制、是否使用統計過程管制監控製程能力等。
Q3:現場參訪時,哪些細節最能看出醫療器材廠商的真實水準?
可觀察作業員是否熟悉流程、現場文件是否為最新版本、設備保養與校正紀錄是否完整,以及物料標示、工具定位與區域劃分是否清楚,觀察現場是否有品質看板展示不良率、客訴數量等即時數據。
Q4:醫療器材廠商在面對多國市場法規時應注意什麼?
醫材上市需符合各市場文件要求,例如美國的 DHR、歐盟 MDR 的技術文件、日本 PMDA 的 Medical Device File,代工廠應能理解不同法規差異,協助準備追溯紀錄、製程證據與文件格式並因應法規更新,才能支援產品進入多國市場。
7.結論:選擇正確的 OEM 夥伴,才能掌握品質競爭力
在高度受法規監管的醫療器材產業中,優質的 OEM 製造商已不再只是加工供應商,而是協助品牌打造安全性、穩定性與市場競爭力的長期合作夥伴。選擇具備 ISO 13485 認證、品質管理系統成熟且能穩定維運的代工廠,雖然初期投入可能較高,但能有效降低召回風險、提升法規審查效率、縮短上市時程,同時強化品牌信譽。
元信豐專注骨科醫材精密加工多年,自 2014 年導入 ISO 13485 以來,持續以國際醫療器材標準作為品質基礎,從原料控管、製程監測、文件追溯到內部訓練與改善,都以最高規格落實。我們相信穩定品質來自制度與文化的累積,而非單一設備或口號。透過完善的品質管理架構,我們協助客戶在產品開發、法規符合性與量產階段取得最佳平衡,並在全球醫療器材市場中建立更具優勢的競爭定位。
8. 立即聯繫元信豐:以更高品質,完成您的醫療器材專案
若您正在尋找值得信賴的醫療器材 OEM 夥伴,或正準備開發新產品、驗證製程、導入量產或面對客戶稽核,我們能提供完整且實務導向的製造支持,協助您建立穩固的品質基礎並加速產品上市,歡迎透過線上表單或來信 sales@ysfbone.com 與我們聯繫,我們將於24小時內回覆,協助您評估需求、提供技術建議,與您共同打造符合國際標準的高品質醫療器材。
9.聲明
本文章內容基於醫療器材產業標準、公開資料與元信豐的實務經驗進行整理,僅供醫材產業與專業人士參考。文中所述的品質管理方法與製造流程,可能因不同產品類別、設計需求、法規要求或製造商條件而有所差異,也不適用於醫療診斷或治療判斷。任何臨床相關決策仍須由具資格的專業醫師進行評估,讀者不應依本文內容進行任何醫療判斷或治療行為。
若企業需導入或改善品質管理系統,建議依據實際產品特性、法規要求與風險等級進行專業評估。本文內容如有未盡之處,歡迎業界夥伴交流指正。
10.參考資料
ISO 13485標準與品質管理
International Organization for Standardization. (2016). ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes.
https://www.iso.org/standard/59752.html
American Society for Quality. (n.d.). What is ISO 13485? Medical Device Quality Management Systems.
https://asq.org/quality-resources/iso-13485
DNV. (n.d.). ISO 13485 – Medical devices quality management.
https://www.dnv.us/services/iso-13485-medical-devices-quality-management-235016/
FMEA分析與風險管理
Institute for Healthcare Improvement. (2023, October 20). Failure Modes and Effects Analysis (FMEA) Tool. https://www.ihi.org/library/tools/failure-modes-and-effects-analysis-fmea-tool
Greenlight Guru. (n.d.). Failure Mode Effects Analysis: What Is It & When Should You Use It?
https://www.greenlight.guru/blog/failure-mode-effects-analysis
National Center for Biotechnology Information. (n.d.). Overview of Failure Mode and Effects Analysis (FMEA): A Patient Safety Tool.
https://pmc.ncbi.nlm.nih.gov/articles/PMC10229026/
VEM Medical. (2025, June 25). Failure Mode and Effects Analysis (FMEA) for Medical Devices.
https://vem-medical.com/fmea-for-medical-devices/
Scilife. (n.d.). What are Failure Modes and Effects Analysis (FMEA)?
https://www.scilife.io/glossary/fmea
IQ/OQ/PQ製程驗證
The FDA Group. (n.d.). A Basic Guide to IQ OQ PQ in FDA Regulated Industries.
https://www.thefdagroup.com/blog/a-basic-guide-to-iq-oq-pq-in-fda-regulated-industries
Device History Record (DHR)
SimplerQMS. (2025, June 30). Device History Record (DHR): Definition, Requirements, and What It Includes.
https://simplerqms.com/device-history-record/
Greenlight Guru. (n.d.). DHF vs. DMR vs. DHR: Differences Explained.
https://www.greenlight.guru/blog/design-history-file-dhf-device-master-record-dmr-device-history-record-dhr
Greenlight Guru. (n.d.). What is Device History Record (DHR)?
https://www.greenlight.guru/glossary/device-history-record
Freyr Solutions. (n.d.). What Is a Device History Record (DHR).
https://www.freyrsolutions.com/what-is-a-device-history-record-dhr
Cognidox. (2022, April 20). DHF, DMR and DHR. Demystifying FDA medical device development requirements.
https://www.cognidox.com/blog/dhf-dmr-dhr
日本 PMDA 與 Medical Device File
Pharmaceuticals and Medical Devices Agency. (2021, March 26). Revision of Japanese Medical Device QMS requirements.
https://www.pmda.go.jp/english/review-services/regulatory-info/0004.html
Emergo by UL. (2024, October). Japan MHLW Ordinance 169 and Medical Device and IVD QMS requirements [White paper].
https://www.emergobyul.com/sites/default/files/2024-10/Japan-MHLW-Ordinance-IVD-QMS-requirements-Whitepaper.pdf
Pharmaceuticals and Medical Devices Agency. (n.d.). Regulations and Approval/Certification of Medical Devices.
https://www.pmda.go.jp/english/review-services/reviews/0004.html
Freyr Solutions. (n.d.). Medical Device Registration in Japan | DMAH Agent & PMDA Support.
https://japan.freyrsolutions.com/medical-devices
TÜV SÜD. (n.d.). PMDA Japan and Medical Device Regulation.
https://www.tuvsud.com/en/industries/healthcare-and-medical-devices/medical-devices-and-ivd/medical-device-market-approval-and-certification/medical-devices-and-compliance-with-japan-pal-regulations
5S管理方法
Wikipedia. (2025, November 30). 5S (methodology).
https://en.wikipedia.org/wiki/5S_(methodology)
Lean Manufacturing Tools. (2015, June 7). What is 5S; Seiri, Seiton, Seiso, Seiketsu, Shitsuke. https://leanmanufacturingtools.org/192/what-is-5s-seiri-seiton-seiso-seiketsu-shitsuke/
Spica. (n.d.). 5S System: The lean way to workplace organization for maximum efficiency. https://www.spica.com/blog/5s-system
Engineers Edge. (n.d.). 5S Methodology - Seiri, Seiton, Seiso, Seiketsu, Shitsuke. https://www.engineersedge.com/manufacturing/5s-methodology.htm
台灣TFDA相關資源
衛生福利部. (2021年4月14日). 醫療器材品質管理系統查核準則 [公告]. 全國法規資料庫. https://law.moj.gov.tw/LawClass/LawAll.aspx?pcode=L0030116
衛生福利部食品藥物管理署. (n.d.). 醫療器材製造業者品質管理系統(QMS)申請.
https://www.fda.gov.tw/Tc/siteContent.aspx?sid=11584
歐盟 MDR 相關資源
European Union. (2017). Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices [Medical Device Regulation].
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
Mantra Systems. (2025, July 22). Post-Market Surveillance (PMS): Understanding PMCF & Vigilance under the EU MDR.
https://mantrasystems.com/articles/pms-understanding-pmcf-vigilance-under-eu-mdr
© 2025 元信豐企業股份有限公司 |骨科植入物及手術器材製造專家、ISO 13485 認證醫材廠、CNC 精密加工 |引用或轉載請註明來源,版權所有。




