In the medical device industry, a robust quality management system is essential for international buyers selecting manufacturing partners. In orthopedic implant development and contract manufacturing, buyers typically inquire about ISO 13485 certification from the outset of the conversation. The industry has clearly shifted from prioritizing the lowest price to demanding consistent quality as global regulations become more stringent.
This article provides a practical overview of ISO 13485 from a manufacturer's perspective. It explains the standard's value, its connection to global regulations, the real-world application of the PDCA cycle, and the challenges manufacturers encounter during implementation. If you are a procurement manager, quality director, or decision-maker evaluating OEM partners, this guide will help you balance supply chain selection, regulatory compliance, and quality assurance.
The medical device industry is undergoing a significant transformation. Previously, companies selected suppliers primarily based on price; however, they now prioritize quality. This shift reflects increasing global awareness of safety and more stringent regulatory requirements.
Industry analysis reveals that approximately 71% of medical device recalls result from manufacturing quality issues rather than design flaws. This is a critical finding. Even with excellent product design, serious problems can arise during production without a stable and verifiable quality system. Such issues can compromise patient safety and damage your brand reputation.
When international brands select OEM partners today, they don't just ask, "Can you make this?" They want to know, "Can you consistently make this correctly?" and "Can you prove it?" That's why ISO 13485 certification is so important in medical device supply chains. It's more than just a certificate on the wall; it demonstrates that a manufacturer has systematic management capabilities and can deliver consistent product quality.
This is especially critical for orthopedic implants. Spinal fixation systems, trauma products, and craniofacial reconstruction devices remain in the human body long-term and endure repeated stress. Even minor manufacturing variations can lead to safety issues during clinical use. Establishing an ISO 13485 quality management system has become the standard requirement for entering international medical device markets.
ISO 13485 is an international quality management system standard specifically designed for medical devices. It assists manufacturers, suppliers, and service providers in establishing a management framework that consistently meets regulatory requirements while ensuring product safety and reliability. The standard encompasses all stages, from medical device design and development to production, installation, and post-market servicing. It serves as a critical compliance benchmark for global medical device markets.
Unlike the general ISO 9001 standard, ISO 13485 specifically focuses on product safety, regulatory compliance, risk management, and traceability. It requires manufacturers to do more than simply satisfy customer needs; they must ensure that products meet safety and regulatory requirements at every stage of the production process. This regulatory-first approach distinguishes the medical device industry from other manufacturing sectors.
2.2 Complete Process Control
In today's stringent regulatory environment, ISO 13485 is more than just a quality certification; it serves as a practical guide for developing quality systems that are both sustainable and verifiable. Its value is evident in several key areas:
2.2.1 Process Stability
ISO 13485 mandates standardized processes to ensure consistent quality across every batch, minimizing variation. This requires clearly defined operating standards and controls at every stage, including raw material inspection, machining parameters, quality checks, and packaging through to shipping.
This stability is especially critical for orthopedic implants. The precision of threads on spinal screws, surface treatment roughness, and material properties must all remain consistent within specified tolerances. Any variation between batches can adversely affect clinical performance.
2.2.2 Record Traceability
Every stage requires comprehensive records that you can verify and trace. This includes raw material sources, process parameters, quality inspections, and shipping details. Maintaining these records is not only essential for regulatory compliance but also forms the foundation of effective risk management. When product issues arise in the market, complete traceability enables you to quickly identify problematic batches, assess their impact, and implement corrective actions.
In practice, each product requires a unique identifier that can be traced back to raw material batch numbers, processing equipment, operators, inspection data, and more. This traceability fosters confidence in quality.
2.2.3 Risk Management
ISO 13485 incorporates risk identification and control from the design phase, preventing potential issues and enhancing product safety and regulatory compliance. This prevention-focused approach mandates risk assessments during product development to identify possible failure modes and implement control measures in manufacturing.
For example, when machining Ti-6Al-4V titanium alloy orthopedic implants, it is essential to evaluate how material hardness influences tool life, how processing temperature impacts material properties, and how surface treatment affects biocompatibility. By conducting a systematic risk analysis, you can implement preventive measures before issues arise.
For medical device brands, these elements are essential for selecting reliable supply chain partners. For OEM manufacturers, they represent the fundamental requirements for building international trust and maintaining competitiveness. As global regulations continue to align with ISO 13485, the medical device industry is progressively moving toward more systematic, process-oriented manufacturing.
2.3 Key Factors in Supply Chain Selection and Competitiveness
For medical device brand owners, these requirements serve as the essential criteria for selecting reliable supply chain partners. For OEM manufacturers, they represent the fundamental threshold for securing international collaborations and enhancing competitiveness. As regulations in various countries increasingly align with ISO 13485, the global medical device industry is evolving toward a more systematic, process-oriented model, with quality management progressively becoming the most critical indicator of manufacturing capability.
ISO 13485 has become the foundation for national regulatory frameworks as the universally accepted quality management standard for medical devices. Major regulatory bodies such as the EU MDR, FDA QMSR, Taiwan TFDA, and Japan PMDA all reference the core principles of ISO 13485 to develop their local quality requirements.
This global standardization offers significant advantages to medical device manufacturers. Choosing manufacturing partners with ISO 13485 certification helps you mitigate compliance risks, reduce review times, streamline technical documentation, and enhance international competitiveness. This allows you to concentrate on product innovation and market expansion.
Must comply with EN ISO 13485:2016 + A11:2021 standards.
Incorporates ISO 13485:2016 by reference
Chapter 2 is aligned with ISO 13485:2016
Certification
A QMS audit must be successfully passed to obtain a manufacturing license.
Requires Notified Body certification
An ISO certificate is optional; however, it must comply with QMSR requirements.
Requires PMDA or RCB QMS assessment
Validity Period
3 years
Typically, 3 years
Ongoing
5 years
Special Needs
Some products are exempt from licensing.
Clinical Evaluation, EUDAMED
Labeling and Packaging Controls
Labeling and Packaging Controls
Table 2: Key Regulatory Differences
3.3 What Each Market Requires
3.3.1 Taiwan TFDA
Taiwan's Medical Device QMS Audit Guidelines were announced on April 14, 2021, and came into effect on May 1, 2021, alongside the Medical Devices Act. Article 2 states that the guidelines adopt ISO 13485:2016 as the audit criteria for manufacturers.
Existing medical device manufacturers must achieve full QMS compliance and obtain manufacturing licenses by April 30, 2024. These manufacturing licenses are valid for a period of three years. Manufacturers are required to apply for re-inspection six to twelve months before the license expiration date.
3.3.2European Union MDR
The EU Medical Device Regulation (EU MDR 2017/745) fully came into effect on May 26, 2021. Article 10.9 mandates that manufacturers establish a quality management system with requirements closely aligned to EN ISO 13485:2016. The 2021 update of EN ISO 13485:2016+A11:2021 introduced Annexes ZA and ZB, integrating the requirements of both the MDR and IVDR with ISO 13485.
EU requirements particularly emphasize clinical evaluation, post-market surveillance, and vigilance systems. Manufacturers must implement comprehensive product lifecycle management. Although the MDR does not explicitly mandate ISO 13485, it is the only QMS standard included in the EU harmonized standards. Compliance with ISO 13485 demonstrates that you meet the fundamental quality requirements for CE marking.
3.3.3 United States FDA QMSR
On January 31, 2024, the FDA published its final rule changing the Quality System Regulation (QSR) to the Quality Management System Regulation (QMSR). Full implementation will occur on February 2, 2026. The QMSR incorporates ISO 13485:2016 by reference into 21 CFR Part 820, fully aligning with international standards.
The final rule mandates compliance with ISO 13485 while incorporating additional requirements from the Food, Drug, and Cosmetic Act. These include expanded design control scope, specific recordkeeping obligations, and enhanced labeling and packaging controls (new Section 820.45). This change officially aligns the U.S. with international standards, which is excellent news for global medical device manufacturers.
3.3.4Japan PMDA
Japan's QMS Ordinance (MHLW Ministerial Ordinance No. 169) was updated on March 26, 2021, to align with ISO 13485:2016, allowing for a three-year transition period. Chapter 2 (Articles 5–64) corresponds to ISO 13485:2016 Articles 4–8, with largely similar requirements, although some minor differences remain.
Chapter 3 introduces additional requirements, particularly the Medical Device File, which differs from the EU MDR technical documentation and more closely resembles the FDA's Device Master Record. The Japanese market places strong emphasis on comprehensive documentation and traceability. Possessing an ISO 13485 certificate alone does not demonstrate compliance with Japan's QMS requirements. It is necessary to fulfill the additional criteria outlined in Ordinance No. 169.
3.4 One System for Multiple Markets
For medical device companies expanding internationally, the most effective strategy is to develop a single, high-standard ISO 13485 quality management system that complies with the requirements of major markets. Here's how:
3.4.1 Documentation Strategy
Implement the most rigorous recordkeeping practices to ensure full traceability. Aim to comply simultaneously with the FDA's Device History Record (DHR), the EU's technical documentation requirements, and Japan's Medical Device File standards. Establish a unified documentation platform capable of managing the varying formats and content requirements across these markets.
3.4.2 Integrated Risk Management
Develop systematic Failure Mode and Effects Analysis (FMEA) processes that comply with ISO 14971 standards. Integrate these processes into every stage of product design and manufacturing. Risk management should not be treated as separate paperwork; instead, it should inform and guide daily decision-making.
3.4.3 Supply Chain Quality
Conduct regular audits and assessments of critical suppliers and maintain an Approved Vendor List (AVL) to ensure supply chain stability. The quality management practices of your suppliers directly impact the compliance of your final product.
3.4.4 Complete Process Validation
Establish comprehensiveIQ/OQ/PQ (Installation, Operational, and Performance Qualification) procedures, particularly for sterilization, specialized processes, and software validation. Process validation is not a one-time event; periodic revalidation is necessary to ensure continued stability.
3.4.5 Post-Market Surveillance
Develop proactive post-market surveillance systems encompassing adverse event reporting, complaint management, vigilance reporting, and Periodic Safety Update Reports (PSURs). This goes beyond mere compliance; it provides essential information for continuous improvement.
This approach requires a higher initial investment but significantly reduces adjustment costs when entering different markets later. It accelerates time-to-market and strengthens your core competitiveness, fostering long-term success in global markets.
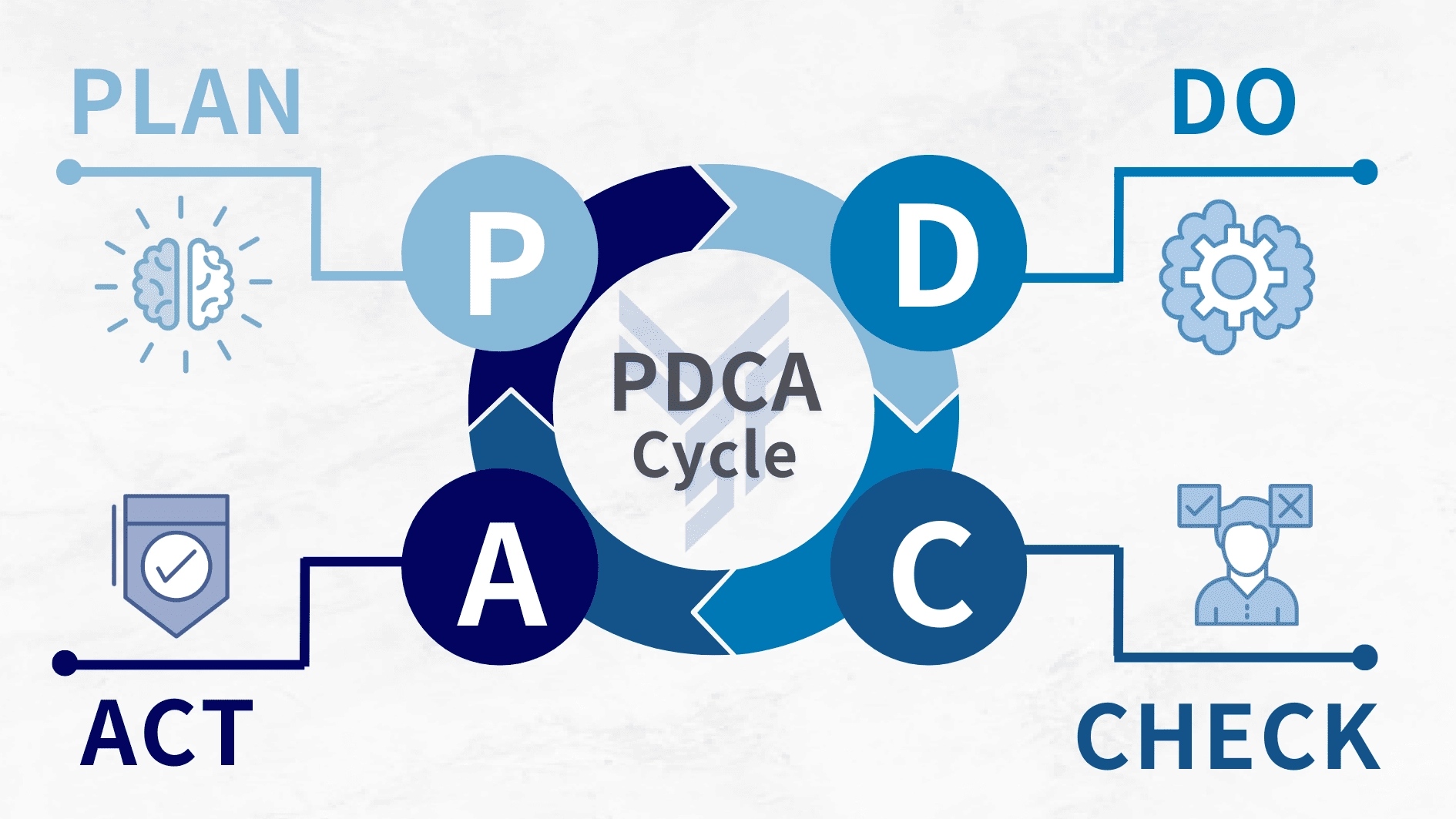
ISO 13485 derives its true value from its foundation on the PDCA cycle (Plan-Do-Check-Act). This approach integrates quality management into daily operations to facilitate continuous improvement. The management philosophy, developed by W. Edwards Deming, emphasizes systematic, ongoing efforts rather than one-time achievements.
4.1.1 Plan
Based on regulatory and customer requirements, establish quality objectives, design process standards, and implement documentation controls. This phase requires clearly defining:
Product Specifications and Acceptance Criteria
Process Parameters and Control Ranges
Inspection Methods and Decision Criteria
Documentation Formats and Management Processes
In orthopedic implant manufacturing, this involves establishing machining parameters for Ti-6Al-4V titanium alloy, specifying surface treatment requirements, and defining dimensional tolerances.
4.1.2 Do
Implement standard operating procedures, train employees, and maintain control over production and work environments. This includes:
Producing according to SOPs
Maintaining and Calibrating Equipment
Recording Process Parameters and Inspection Data
Maintaining Environmental Controls in Cleanrooms
In practice, this requires comprehensive work instructions and ensuring that every operator receives adequate training and assessment.
4.1.3 Check
Regularly monitor process and quality indicators, conduct internal audits, collect data, and ensure compliance. This includes:
In-Process Quality Control
Final Quality Control
Internal Audits
Management Reviews
Through data analysis, you can detect process drift, equipment malfunctions, or human errors early and take action before these issues escalate.
4.1.4 Act Based on the check results, analyze the findings, implement corrective and preventive actions, and optimize the system. This includes:
Root Cause Analysis of Nonconforming Products
Implementing Corrective Actions
Setting Up Preventive Actions
Updating Documentation and Operating Standards
4.2 PDCA in Real Manufacturing
Here is how the PDCA cycle works, using spinal fixation screw production as an example:
Plan Phase: Establish thread machining tool paths, cutting parameters, coolant formulation, and thread pitch tolerance at ±0.02 mm.
Do Phase:The machining phase, according to the specified parameters, recording each batch's machining time, tool wear, and surface roughness.
Check Phase: Use 2.5D image measurement systems to verify thread pitch precision. Monitor for drift trends beginning with batch 15.
Act Phase: After analysis revealed that the root cause was tool wear, the tool replacement cycle was adjusted from every 20 batches to every 50 batches. This change immediately resolved the deviation issue, and the standard operating procedures were updated accordingly.
This demonstrates that PDCA cycles are not focused on fixing problems after they occur. Instead, they employ continuous monitoring and data analysis to address issues before they develop into defects. This proactive management approach is the core principle of ISO 13485.
During the implementation of ISO 13485, medical device manufacturers encounter numerous challenges at both organizational and technical levels. However, by employing effective strategies and tools, these challenges can be transformed into opportunities to improve quality capabilities and enhance competitiveness.
5.1 Documentation Management
ISO 13485 mandates comprehensive documentation systems, including quality manuals, procedures, work instructions, and record forms. For many small and medium-sized manufacturers, this requirement can be overwhelming.
Challenge: Managing an excessive number of documents leads to version control issues, hampers cross-departmental collaboration, and results in time-consuming updates prone to numerous errors.
Solution: Implement Document Management Systems (DMS) to create electronic document libraries featuring permission controls and version tracking. Designate document management leads to oversee and regularly assess the effectiveness of documentation. Additionally, utilize cloud collaboration platforms to enhance communication across departments.
5.2 Process Validation Resources
ISO 13485 requires the validation of critical processes to ensure they consistently produce products that meet specifications. This needs IQ/OQ/PQ validation, which demands significant time and resources.
Challenges include complex validation procedures, the need for statistical analysis skills, potential production disruptions, and a shortage of specialists.
Solution: Develop validation plan templates and implement validation in phases. Begin with high-risk processes, then gradually extend to others. Engage external consultants as needed to accelerate the process. Simultaneously, cultivate internal talent to strengthen validation capabilities.
5.3 Supply Chain Management
Medical device manufacturing involves multiple suppliers for raw materials, components, and outsourced services. All of these must be included within your quality management scope.
Challenge: Inconsistent supplier quality, high communication costs, difficulty controlling upstream quality, and insufficient supply chain transparency.
Solution: Maintain an Approved Vendor List (AVL). Conduct regular supplier audits and performance evaluations. Require critical suppliers to provide quality documentation. Perform on-site inspections when necessary. Establish tiered supplier management with varying controls for each tier.
5.4 Training Employees and Cultivating Organizational Culture
ISO 13485 ultimately depends on individuals' understanding and effectively implementing quality management.
Challenge: Employees are unfamiliar with ISO requirements, merely going through the motions, lacking motivation for improvement, and demonstrating insufficient quality awareness.
Solution: Develop comprehensive training programs that cover the fundamentals of ISO 13485, operational skills, and quality awareness. Utilize internal audits to identify issues and link the results of improvements to performance metrics to foster a culture of quality. Additionally, organize regular quality improvement competitions to encourage active participation.
5.5 Maintaining Momentum
Achieving ISO 13485 certification is only the beginning. Maintaining the system's effectiveness and driving continuous improvement represent the ongoing challenges.
Challenge: After certification, complacency sets in; the system becomes rigid, motivation for improvement wanes, and processes are maintained solely to satisfy audit requirements.
Solution: Establish annual quality objectives and KPIs. Run regular management reviews to assess the status of the quality management system. Encourage employee suggestions for improvement by implementing a rewards program. Participate in industry forums to learn and adopt best practices. Align quality management with business objectives, transforming quality into a competitive advantage rather than a burden.
Since obtaining ISO 13485 certification in 2014 and officially becoming a medical device manufacturer, YSF MEDICAL has focused on developing quality management systems that comply with international standards. We are not merely seeking certification; we are establishing systems that genuinely enhance product quality, mitigate risks, and support customer needs.
6.1 Comprehensive Quality Control
Every stage has clear control standards and documentation, from product design interfaces and raw material inspections to process parameters, quality checks, packaging, shipping, and after-sales service. We utilize precision inspection equipment, such as 2.5D image measuring systems, to ensure that every orthopedic implant meets specifications.
6.2 Risk Management
For each product line, we conduct systematic risk assessments, identify potential failure modes, and implement corresponding controls within our processes. For example, during the machining of Ti-6Al-4V titanium alloy, we employ patented cooling technology to reduce machining temperatures and prevent degradation of material properties.
6.3 Documentation Traceability
We maintain comprehensive product histories, including raw material batch numbers, machining parameters, inspection data, and shipping records. All information is fully traceable. When customers require audits or market surveillance authorities request documentation, we can promptly provide complete records.
6.4 Cultivating a Culture of Continuous Improvement
We regularly conduct internal audits and management reviews, monitor system status, and encourage employees to submit improvement suggestions. Through PDCA cycles, we continuously optimize processes, enhance efficiency, and reduce defect rates.
As a proven one-stop medical device contract manufacturing partner, we assist customers through everything from sample prototyping and process implementation to technical documentation preparation and third-party audit support. We serve high-standard markets, including the US, EU, and Japan.
1. What is ISO 13485, and why is it essential for the medical device industry?
ISO 13485 is a quality management system standard specifically designed for the medical device industry to ensure that products remain safe, effective, and compliant with regulatory requirements throughout their entire lifecycle. It encompasses design, development, production, packaging, sterilization, storage, distribution, and post-market activities, serving as a critical framework adopted by regulatory authorities worldwide. Implementing ISO 13485 not only strengthens quality consistency but also enhances traceability, risk management capabilities, and documentation transparency. For medical device manufacturers, ISO 13485 is an essential benchmark for gaining international customer trust and accessing global supply chains. Through comprehensive system management, companies can reduce defect rates, shorten customer audit times, and improve market competitiveness.
2.What are the differences between ISO 13485 and ISO 9001? Which standard is more important for the medical industry?
ISO 9001 is a general quality management system applicable across all industries, whereas ISO 13485 is specifically designed for the medical device sector, emphasizing regulatory compliance, product safety, and risk management. The most significant difference lies in their management philosophies: ISO 13485 adopts a "regulation-first" approach, requiring companies to establish processes in accordance with national regulatory requirements, such as traceability management, risk control, design validation, and process validation. In contrast, ISO 9001 focuses on achieving customer satisfaction, while ISO 13485 prioritizes product safety during clinical use. For medical device manufacturers, ISO 13485 is an essential core standard that helps companies comply with global regulatory requirements and enhances opportunities for collaboration with international brands.
3.Does having ISO 13485 certification mean that products can be sold in the US, Europe, or Japan?
ISO 13485 is the most internationally recognized quality management system for medical devices; however, it is not a product market authorization and cannot replace country-specific regulatory approvals. For example, the U.S. market still requires FDA 510(k) clearance or PMA approval, the EU mandates CE technical documentation review, and Japan requires PMDA approval. The purpose of ISO 13485 is to ensure that companies have quality management systems compliant with regulations, thereby reducing issues during the review process and increasing the likelihood that technical documentation will pass evaluation. Therefore, ISO 13485 serves as a foundational threshold, while actual market entry requires obtaining the appropriate product certifications for each specific market.
4.How long does ISO 13485 certification take, and what steps should be taken after obtaining it?
The timeline for implementing ISO 13485 varies depending on the company's size and system maturity, typically ranging from six months to one year. The process involves establishing procedures, preparing documentation, managing risks, validating processes, conducting training, and performing internal audits. After obtaining certification, companies must undergo annual surveillance audits and continuously update documentation, improve processes, maintain traceability, and manage risks. The essence of ISO 13485 lies in the PDCA (Plan-Do-Check-Act) cycle of continuous improvement; thus, certification is not the endpoint but rather a commitment to consistently maintain the quality system, reduce market risks, and respond to regulatory updates. Stable, long-term operation is what truly enables the ISO system to deliver value.
5.How can you verify the authenticity of an ISO 13485 certificate? How can you mitigate partnership risks?
Methods to verify the authenticity of an ISO 13485 certificate include confirming whether the certification body listed on the certificate is an internationally recognized third party, such as SGS, BSI, DNV, or TÜV. You can visit the issuing body's website directly to verify the certificate number and check whether the certificate scope covers the relevant manufacturing activities and product categories. If a manufacturer is willing to provide external audit reports, records of nonconformities, and corrective actions, this also indicates a more mature quality system. The most reliable method is to conduct an on-site audit to confirm whether the company's daily operations align with documented procedures. Manufacturers with greater transparency typically have more trustworthy quality systems.
In the medical device industry, ISO 13485 has evolved from a nice-to-have to an essential requirement. It is more than just a gateway to international markets; it is crucial for establishing a reputation for quality and maintaining competitiveness. For brands, selecting OEM partners with ISO 13485 certification reduces supply chain risks and ensures consistent product quality. Although implementing ISO 13485 requires significant resources for manufacturers, the long-term benefits far outweigh the costs.
ISO 13485 offers more than just standards. It encompasses systematic process management, comprehensive record traceability, and continuous improvement mechanisms, embodying a management philosophy. This standard helps manufacturers transition from reactive inspection to proactive prevention, from experience-based approaches to data-driven decision-making, and from reliance on individuals to dependence on robust systems.
YSF MEDICAL has specialized in precision machining of orthopedic medical devices for over 30 years, establishing comprehensive quality management systems based on the ISO 13485 standard. We not only deliver products that comply with international standards but also assist customers in optimizing processes, enhancing quality performance, and strengthening regulatory compliance and global deployment strategies.
Looking for a trusted medical device OEM partner? Contact YSF MEDICAL. With our extensive manufacturing experience, comprehensive quality systems, and professional technical teams, we will help you succeed in the global medical device market.
📩 Get in touch today: Fill out our form or email us at sales@ysfbone.com. We will respond within 24 hours.
This content is intended for reference by medical professionals and the healthcare industry. Some information is sourced from publicly available materials or expert opinions and may be incomplete or require further verification. Feedback and professional discussion are encouraged.
Important Reminder: Any medical diagnosis or treatment decisions should be based exclusively on the professional judgment of qualified clinicians. Patients should not make medical decisions solely on the information provided in this document.
ISO 13485 Standard and Resources
International Organization for Standardization. (2016). ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes. https://www.iso.org/standard/59752.html
U.S. Food and Drug Administration. (2011). Process validation: General principles and practices [Guidance for industry]. https://www.fda.gov/media/71021/download
Taiwan Food and Drug Administration, Ministry of Health and Welfare. (n.d.). Application for Quality Management System (QMS) for Medical Device Manufacturers. https://www.fda.gov.tw/Tc/siteContent.aspx?sid=11584
Plastics Industry Development Center. (n.d.). Republic of China: Guidance for Registration of Domestic Medical Device Quality Management Systems. https://www.pidc.org.tw/materials.php?id=830
Quality Management Tools and Methods
American Society for Quality. (n.d.). What is Failure Mode and Effects Analysis (FMEA)? https://asq.org/quality-resources/fmea
Industry Resources and Research
Peters, W., Pellerin, C., & Janney, C. (2020). Analysis: Using the FDA MAUDE and Medical Device Recall databases to design better devices. Biomedical Instrumentation & Technology, 54(3), 178-194. https://doi.org/10.2345/0899-8205-54.3.178